Microbial Community Composition and Antibiotic Resistance Genes within a North Carolina Urban Water System

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
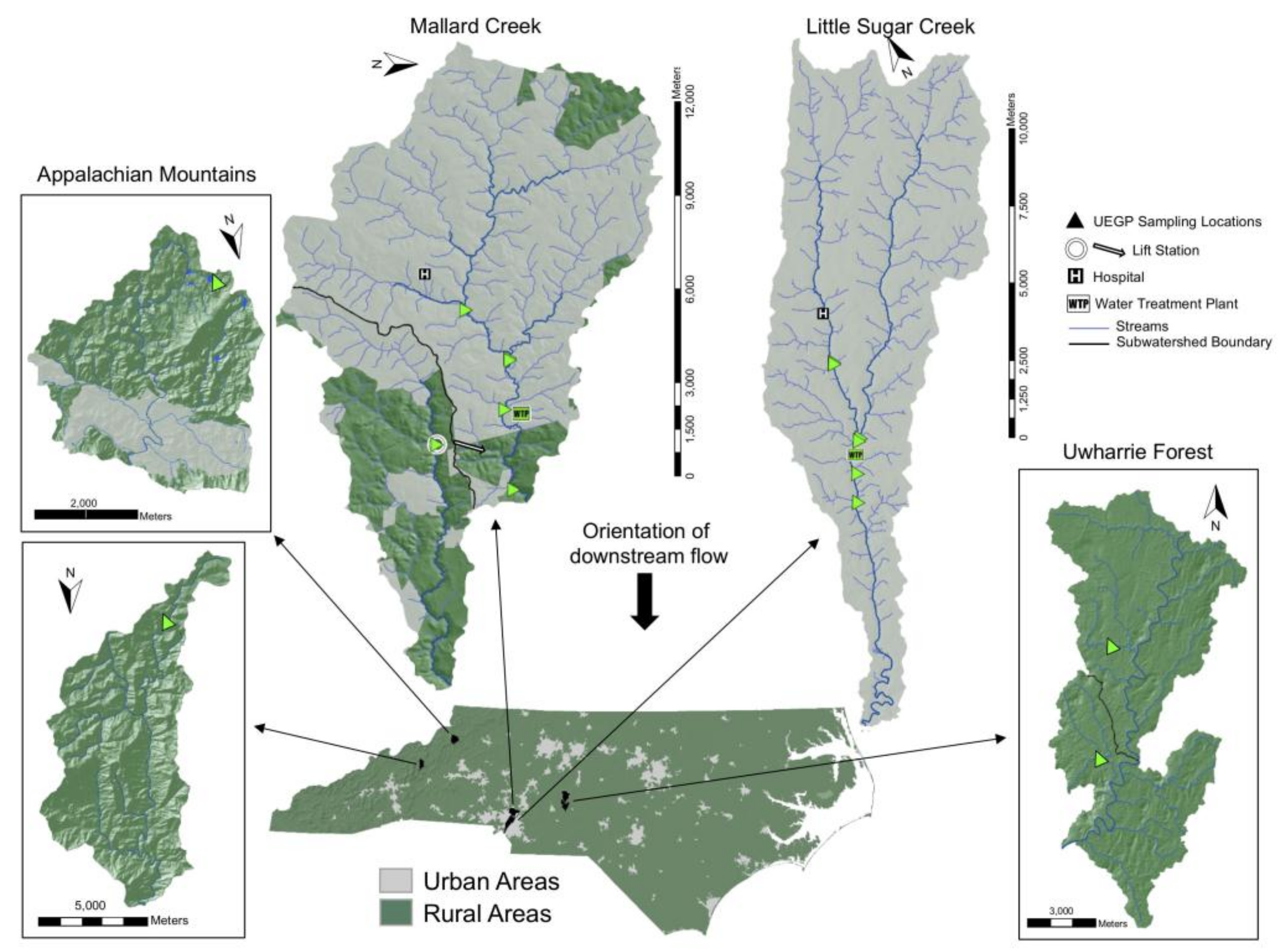
2.1. Selection of Sampling Sites
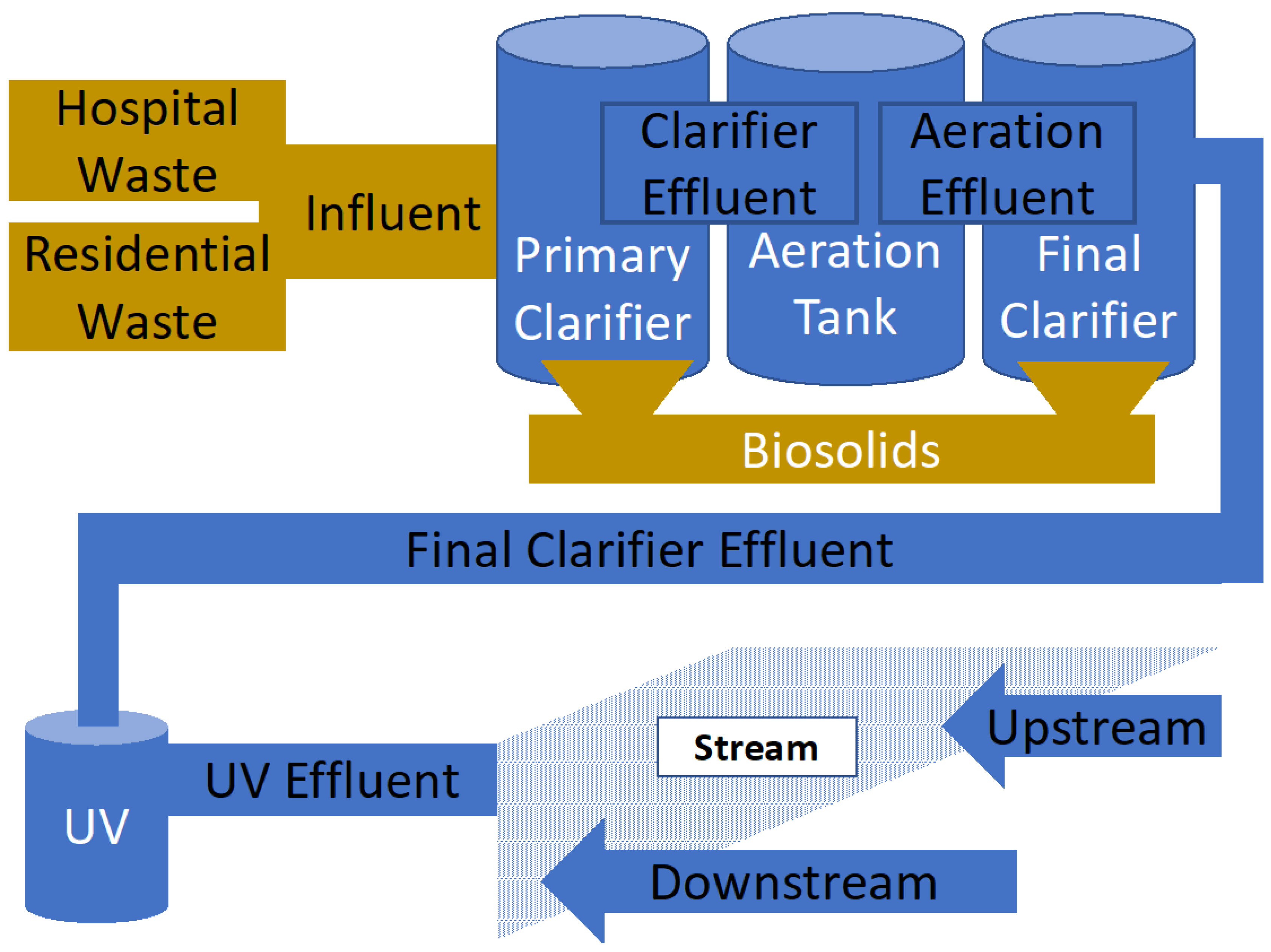
2.2. Treatment Methods and Conditions in Charlotte Water Facilities
2.3. Sample Collection and Handling
2.4. Detection of Antibiotic Compounds by Mass Spectrometry
2.5. DNA Extraction
2.6. Shotgun Metagenomic Library Preparation and Sequencing
2.7. DNA Sequence Trimming and Quality Control
2.8. Metagenomic Classification Analysis Using MetaPhlAn2
2.9. Identification and Quantitation of Resistance Elements Using ShortBRED
2.10. Functional Classification Analysis Using HUMAnN2
2.11. Statistical Methods
3. Results
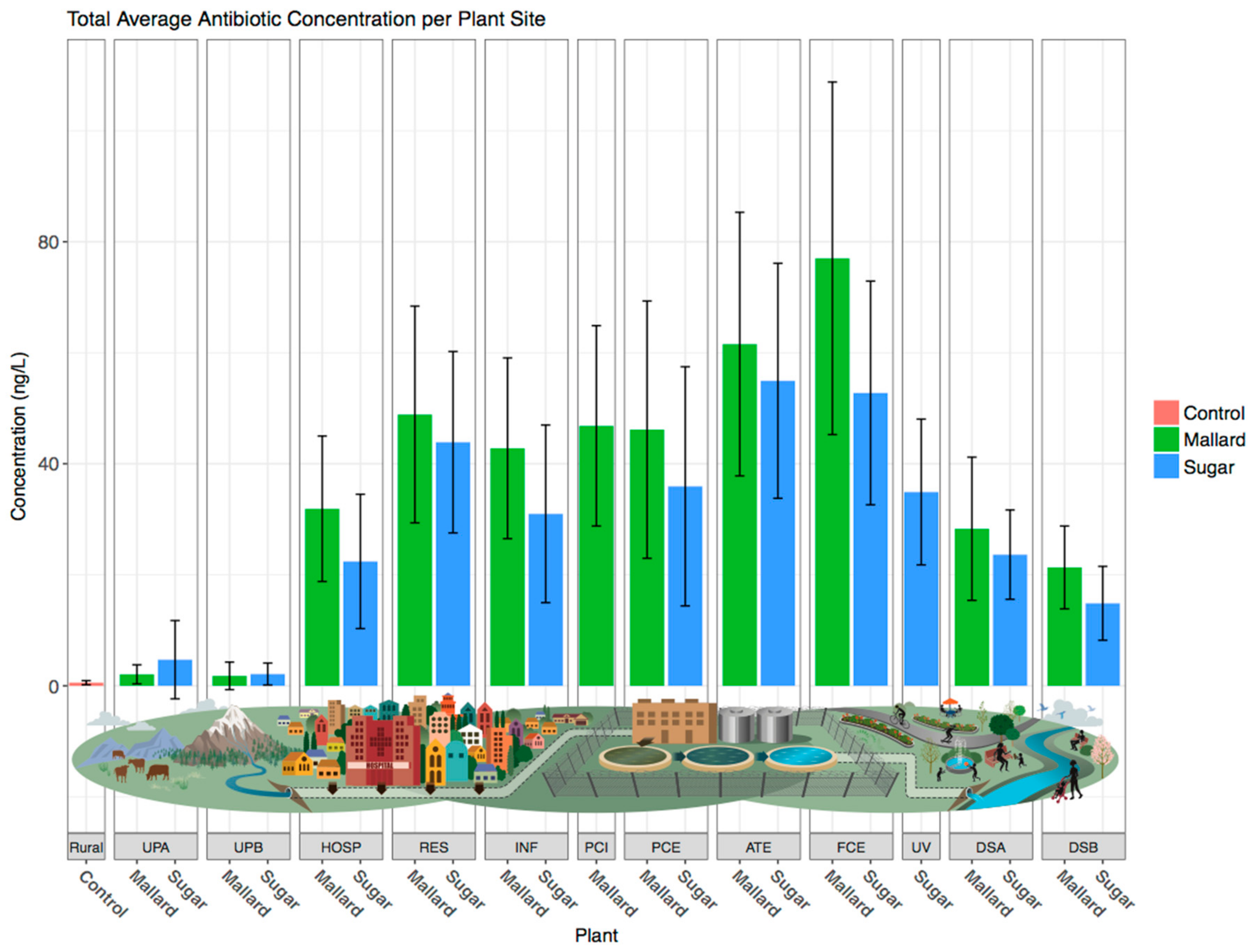
3.1. Antibiotic Concentrations Are Elevated Downstream of Wastewater Treatment Plants
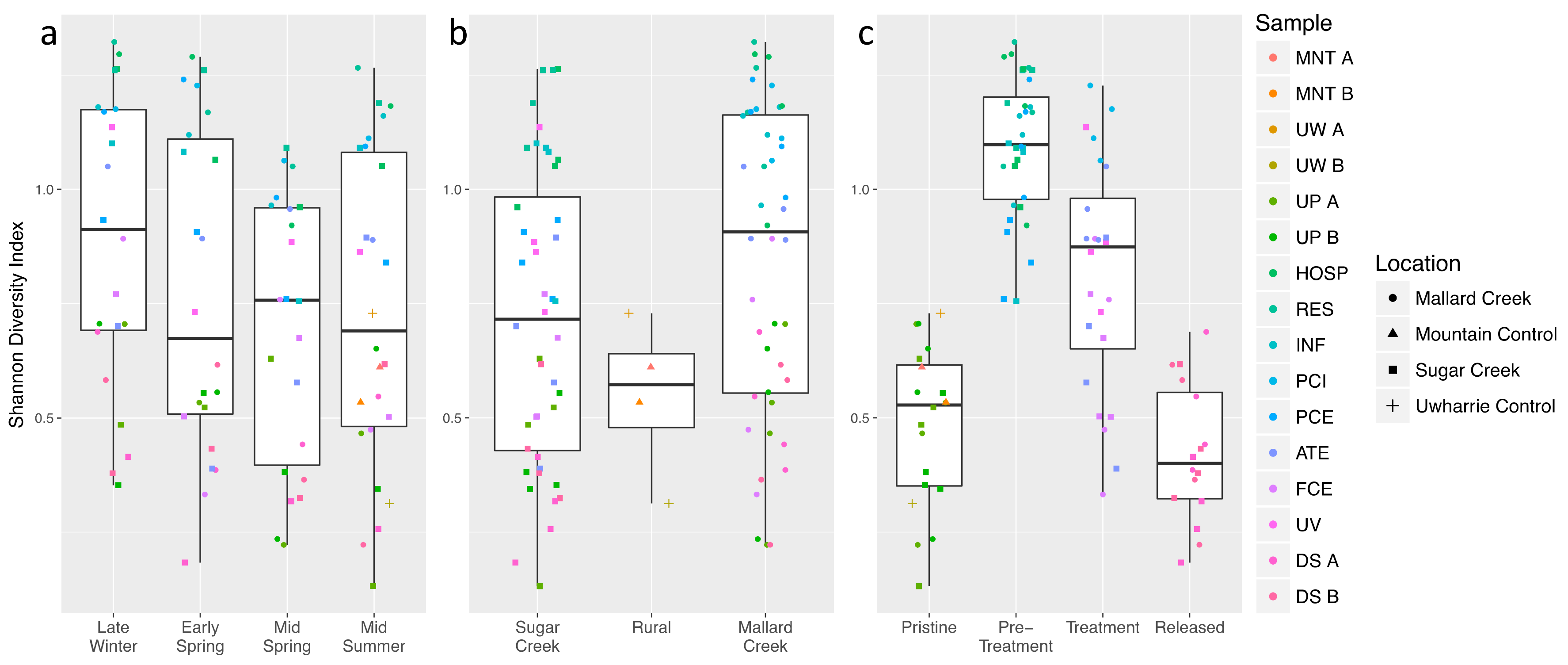
3.2. Treated Wastewater Microbial Communities Become More like Fresh Water Communities as Waste Progresses through the Treatment Process
3.3. Microbial Community Shifts during Wastewater Processing
3.4. Shifts in Stream Microbial Community Function Are Observed Downstream of Treated Wastewater Release Points
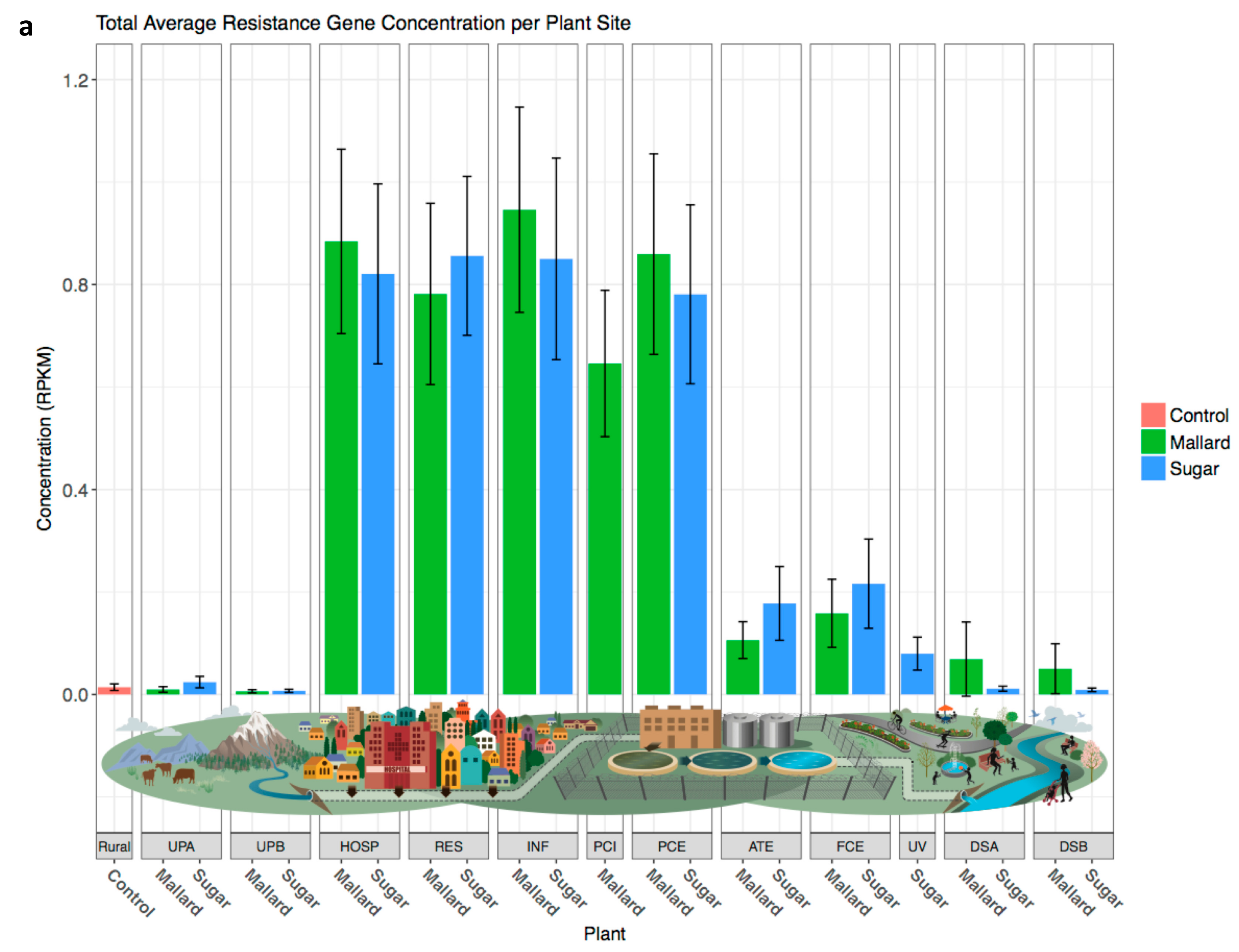
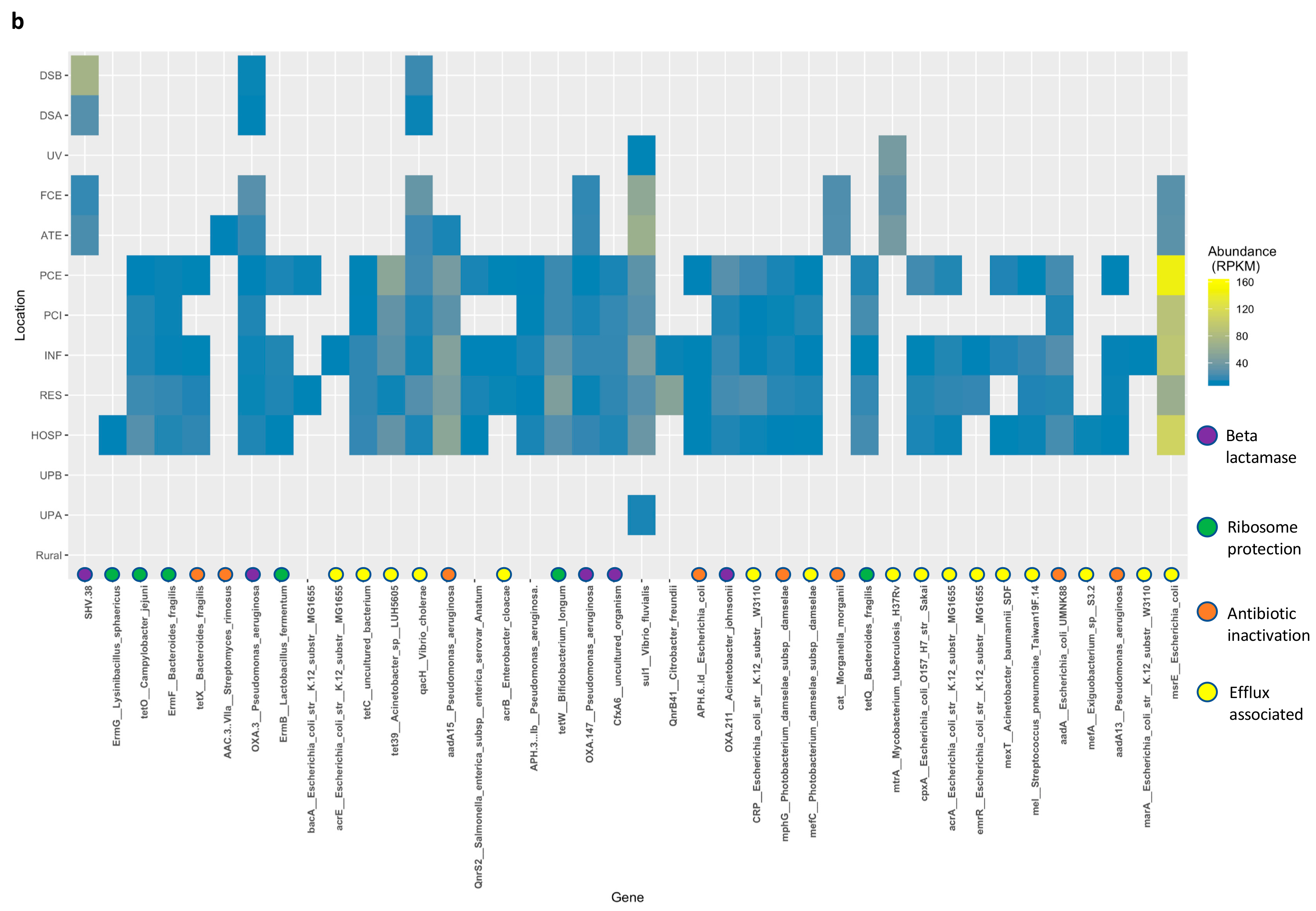
3.5. Resistance Genes Are Slightly More Abundant Downstream of Wastewater Treatment Plants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walsh, C.J.; Roy, A.H.; Feminella, J.W.; Cottingham, P.D.; Groffman, P.M.; Morgan, R.P., II. The urban stream syndrome: Current knowledge and the search for a cure. J. N. Am. Benthol. Soc. 2005, 24, 706–723. [Google Scholar] [CrossRef]
- Yu, S.; Wu, Q.; Li, Q.; Gao, J.; Lin, Q.; Ma, J.; Xu, Q.; Wu, S. Anthropogenic land uses elevate metal levels in stream water in an urbanizing watershed. Sci. Total Environ. 2014, 488–489, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.C.; Newton, R.J.; Dila, D.K.; McLellan, S.L. Urban microbial ecology of a freshwater estuary of Lake Michigan. Elementa 2015, 3, 000064. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Ren, Z.; Zhang, H.; Zhang, M.; Zhang, Y.; Liu, X.; Peng, W. Influences of anthropogenic land use on microbial community structure and functional potentials of stream benthic biofilms. Sci. Rep. 2017, 7, 15117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, R.J.; McLellan, S.L. A unique assemblage of cosmopolitan freshwater bacteria and higher community diversity differentiate an urbanized estuary from oligotrophic Lake Michigan. Front. Microbiol. 2015, 6, 1028. [Google Scholar] [CrossRef] [PubMed]
- McLellan, S.L.; Fisher, J.C.; Newton, R.J. The microbiome of urban waters. Int. Microbiol. 2015, 18, 141–149. [Google Scholar] [PubMed]
- Kasprzyk-Hordern, B.; Dinsdale, R.M.; Guwy, A.J. The removal of pharmaceuticals, personal care products, endocrine disruptors and illicit drugs during wastewater treatment and its impact on the quality of receiving waters. Water Res. 2009, 43, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Niemuth, N.J.; Jordan, R.; Crago, J.; Blanksma, C.; Johnson, R.; Klaper, R.D. Metformin exposure at environmentally relevant concentrations causes potential endocrine disruption in adult male fish. Environ. Toxicol. Chem. 2015, 34, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Conley, J.M.; Evans, N.; Mash, H.; Rosenblum, L.; Schenck, K.; Glassmeyer, S.; Furlong, E.T.; Kolpin, D.W.; Wilson, V.S. Comparison of in vitro estrogenic activity and estrogen concentrations in source and treated waters from 25 U.S. Drinking water treatment plants. Sci. Total Environ. 2017, 579, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Beresford, N.; Baynes, A.; Kanda, R.; Mills, M.R.; Arias-Salazar, K.; Collins, T.J.; Jobling, S. Use of a battery of chemical and ecotoxicological methods for the assessment of the efficacy of wastewater treatment processes to remove estrogenic potency. J. Vis. Exp. 2016, 54243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, T.; Guo, C.; Lv, J.; Hua, Z.; Hou, S.; Zhang, Y.; Meng, W.; Xu, J. Drugs of abuse and their metabolites in the urban rivers of Beijing, China: Occurrence, distribution, and potential environmental risk. Sci. Total Environ. 2017, 579, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Sheng, G.P.; Lu, Y.Z.; Zeng, R.J.; Yu, H.Q. Removal of antibiotic resistance genes from wastewater treatment plant effluent by coagulation. Water Res. 2017, 111, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Prasse, C.; Schlüsener, M.P.; Schulz, R.; Ternes, T.A. Antiviral drugs in wastewater and surface waters: A new pharmaceutical class of environmental relevance? Environ. Sci. Technol. 2010, 44, 1728–1735. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Soares, A.D.; Adejumo, H.; McDiarmid, M.; Squibb, K.; Blaney, L. Detection of a wide variety of human and veterinary fluoroquinolone antibiotics in municipal wastewater and wastewater-impacted surface water. J. Pharm. Biomed. Anal. 2015, 106, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D. Evolution of antibiotic resistance at non-lethal drug concentrations. Drug Resist. Updates 2012, 15, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.; Andersson, D.I. Selection of resistance at lethal and non-lethal antibiotic concentrations. Curr. Opin. Microbiol. 2012, 15, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Xia, Y.; Li, B.; Yang, Y.; Li, L.-G.; Tiedje, J.M.; Zhang, T. Metagenomic assembly reveals hosts of antibiotic resistance genes and the shared resistome in pig, chicken, and human feces. Environ. Sci. Technol. 2016, 50, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Call, D.R.; Matthews, L.; Subbiah, M.; Liu, J. Do antibiotic residues in soils play a role in amplification and transmission of antibiotic resistant bacteria in cattle populations? Front. Microbiol. 2013, 4, 193. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-H.; Qiao, M.; Su, J.-Q.; Chen, Z.; Zhou, X.; Zhu, Y.-G. High throughput profiling of antibiotic resistance genes in urban park soils with reclaimed water irrigation. Environ. Sci. Technol. 2014, 48, 9079–9085. [Google Scholar] [CrossRef] [PubMed]
- Fahrenfeld, N.; Ma, Y.; O’Brien, M.; Pruden, A. Reclaimed water as a reservoir of antibiotic resistance genes: Distribution system and irrigation implications. Front. Microbiol. 2013, 4, 130. [Google Scholar] [CrossRef] [PubMed]
- Pruden, A. Balancing water sustainability and public health goals in the face of growing concerns about antibiotic resistance. Environ. Sci. Technol. 2014, 48, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Pehrsson, E.C.; Tsukayama, P.; Patel, S.; Mejia-Bautista, M.; Sosa-Soto, G.; Navarrete, K.M.; Calderon, M.; Cabrera, L.; Hoyos-Arango, W.; Bertoli, M.T.; et al. Interconnected microbiomes and resistomes in low-income human habitats. Nature 2016, 533, 212–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaPara, T.M.; Burch, T.R.; McNamara, P.J.; Tan, D.T.; Yan, M.; Eichmiller, J.J. Tertiary-treated municipal wastewater is a significant point source of antibiotic resistance genes into duluth-superior harbor. Environ. Sci. Technol. 2011, 45, 9543–9549. [Google Scholar] [CrossRef] [PubMed]
- Munck, C.; Albertsen, M.; Telke, A.; Ellabaan, M.; Nielsen, P.H.; Sommer, M.O. Limited dissemination of the wastewater treatment plant core resistome. Nat. Commun. 2015, 6, 8452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christgen, B.; Yang, Y.; Ahammad, S.Z.; Li, B.; Rodriquez, D.C.; Zhang, T.; Graham, D.W. Metagenomics shows that low-energy anaerobic–aerobic treatment reactors reduce antibiotic resistance gene levels from domestic wastewater. Environ. Sci. Technol. 2015, 49, 2577–2584. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Waldron, L.; Ballarini, A.; Narasimhan, V.; Jousson, O.; Huttenhower, C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 2012, 9, 811–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Department Core Team. R: A Language and Environment for Statistical Computing; The R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Kaminski, J.; Gibson, M.K.; Franzosa, E.A.; Segata, N.; Dantas, G.; Huttenhower, C. High-specificity targeted functional profiling in microbial communities with shortbred. PLoS Comput. Biol. 2015, 11, e1004557. [Google Scholar] [CrossRef] [PubMed]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H.; the UniProt, C. Uniref clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar]
- Wang, M.; Shen, W.; Yan, L.; Wang, X.H.; Xu, H. Stepwise impact of urban wastewater treatment on the bacterial community structure, antibiotic contents, and prevalence of antimicrobial resistance. Environ. Pollut. 2017, 231, 1578–7585. [Google Scholar] [CrossRef] [PubMed]
- Aubertheau, E.; Stalder, T.; Mondamert, L.; Ploy, M.C.; Dagot, C.; Labanowski, J. Impact of wastewater treatment plant discharge on the contamination of river biofilms by pharmaceuticals and antibiotic resistance. Sci. Total Environ. 2017, 579, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Y.; Zhao, L.; Li, Y.; Xie, S.; Liu, Y. Distribution of sediment bacterial and archaeal communities in plateau freshwater lakes. Appl. Microbiol. Biotechnol. 2015, 99, 3291–3302. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, W.; Liu, R.; Li, Q.; Li, B.; Wang, S.; Song, C.; Qiao, C.; Mulchandani, A. Phylogenetic diversity and metabolic potential of activated sludge microbial communities in full-scale wastewater treatment plants. Environ. Sci. Technol. 2011, 45, 7408–7415. [Google Scholar] [CrossRef] [PubMed]
- McLellan, S.L.; Huse, S.M.; Mueller-Spitz, S.R.; Andreishcheva, E.N.; Sogin, M.L. Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ. Microbiol. 2010, 12, 378–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacRae, J.D.; Smit, J. Characterization of caulobacters isolated from wastewater treatment systems. Appl. Environ. Microbiol. 1991, 57, 751–758. [Google Scholar] [PubMed]
- Lücker, S.; Wagner, M.; Maixner, F.; Pelletier, E.; Koch, H.; Vacherie, B.; Rattei, T.; Damsté, J.S.S.; Spieck, E.; Le Paslier, D.; et al. A metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc. Nat. Acad. Sci. USA 2010, 107, 13479. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Choi, K.M.; Yin, C.R.; Lee, K.Y.; Im, W.T.; Lim, J.H.; Lee, S.T. Odorous swine wastewater treatment by purple non-sulfur bacteria, Rhodopseudomonas palustris, isolated from eutrophicated ponds. Biotechnol. Lett. 2004, 26, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Martinez, A.; Sihvonen, M.; Muñoz-Palazon, B.; Rodriguez-Sanchez, A.; Mikola, A.; Vahala, R. Microbial ecology of full-scale wastewater treatment systems in the polar arctic circle: Archaea, bacteria and fungi. Sci. Rep. 2018, 8, 2208. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.M.; Albertsen, M.; Vollertsen, J.; Nielsen, P.H. The activated sludge ecosystem contains a core community of abundant organisms. ISME J 2016, 10, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Balcom, I.N.; Driscoll, H.; Vincent, J.; Leduc, M. Metagenomic analysis of an ecological wastewater treatment plant’s microbial communities and their potential to metabolize pharmaceuticals. F1000Research 2016, 5, 1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medhi, K.; Thakur, I.S. Bioremoval of nutrients from wastewater by a denitrifier paracoccus denitrificans istod1. Bioresour. Technol. Rep. 2018, 1, 56–60. [Google Scholar] [CrossRef]
- Pagnier, I.; Croce, O.; Robert, C.; Raoult, D.; La Scola, B. Genome sequence of Reyranella massiliensis, a bacterium associated with amoebae. J. Bacteriol. 2012, 194, 5698. [Google Scholar] [CrossRef] [PubMed]
- Pehl, M.J.; Jamieson, W.D.; Kong, K.; Forbester, J.L.; Fredendall, R.J.; Gregory, G.A.; McFarland, J.E.; Healy, J.M.; Orwin, P.M. Genes that influence swarming motility and biofilm formation in Variovorax paradoxus eps. PLoS ONE 2012, 7, e31832. [Google Scholar] [CrossRef] [PubMed]
- Kampfer, P.; Witzenberger, R.; Denner, E.B.; Busse, H.J.; Neef, A. Sphingopyxis witflariensis sp. Nov., isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 2002, 52, 2029–2034. [Google Scholar] [PubMed]
- Zielinska, M.; Rusanowska, P.; Jarzabek, J.; Nielsen, J.L. Community dynamics of denitrifying bacteria in full-scale wastewater treatment plants. Environ. Technol. 2016, 37, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Ju, F.; Zhang, T. Tracking human sewage microbiome in a municipal wastewater treatment plant. Appl. Microbiol. Biotechnol. 2014, 98, 3317–3326. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Sanseverino, J.; Chauhan, A.; Lucas, S.; Copeland, A.; Lapidus, A.; Del Rio, T.G.; Dalin, E.; Tice, H.; Bruce, D.; et al. Complete genome sequence of Thauera aminoaromatica strain mz1t. Stand. Genom. Sci. 2012, 6, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Al Atrouni, A.; Joly-Guillou, M.L.; Hamze, M.; Kempf, M. Reservoirs of non-baumannii acinetobacter species. Front. Microbiol. 2016, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Chagas, T.P.; Seki, L.M.; Cury, J.C.; Oliveira, J.A.; Davila, A.M.; Silva, D.M.; Asensi, M.D. Multiresistance, beta-lactamase-encoding genes and bacterial diversity in hospital wastewater in rio de janeiro, brazil. J. Appl. Microbiol. 2011, 111, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Carr, E.L.; Eales, K.L.; Seviour, R.J. Substrate uptake by Gordonia amarae in activated sludge foams by fish-mar. Water Sci. Technol. 2006, 54, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Hanada, S.; Liu, W.T.; Shintani, T.; Kamagata, Y.; Nakamura, K. Tetrasphaera elongata sp. Nov., a polyphosphate-accumulating bacterium isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 2002, 52, 883–887. [Google Scholar] [PubMed]
- Zhang, B.; Xu, X.; Zhu, L. Structure and function of the microbial consortia of activated sludge in typical municipal wastewater treatment plants in winter. Sci. Rep. 2017, 7, 17930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abubucker, S.; Segata, N.; Goll, J.; Schubert, A.M.; Izard, J.; Cantarel, B.L.; Rodriguez-Mueller, B.; Zucker, J.; Thiagarajan, M.; Henrissat, B.; et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol. 2012, 8, e1002358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A.; et al. The metacyc database of metabolic pathways and enzymes and the biocyc collection of pathway/genome databases. Nucleic Acids Res. 2014, 42, D459–D471. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D. Microbiological effects of sublethal levels of antibiotics. Nat. Rev. Microbiol. 2014, 12, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Kolpin, D.W.; Furlong, E.T.; Meyer, M.T.; Thurman, E.M.; Zaugg, S.D.; Barber, L.B.; Buxton, H.T. Pharmaceuticals, hormones, and other organic wastewater contaminants in U.S. Streams, 1999–2000: A national reconnaissance. Environ. Sci. Technol. 2002, 36, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rojas, A.; Rodriguez-Beltran, J.; Couce, A.; Blazquez, J. Antibiotics and antibiotic resistance: A bitter fight against evolution. Int. J. Med. Microbiol. 2013, 303, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.J.; McLellan, S.L.; Dila, D.K.; Vineis, J.H.; Morrison, H.G.; Eren, A.M.; Sogin, M.L. Sewage reflects the microbiomes of human populations. MBio 2015, 6, e02574. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Chung, H.J.; Wen Di, D.Y.; Dodd, M.C.; Hur, H.G.; Lee, Y. Inactivation efficiency of plasmid-encoded antibiotic resistance genes during water treatment with chlorine, uv, and uv/h2o2. Water Res. 2017, 123, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Laquaz, M.; Dagot, C.; Bazin, C.; Bastide, T.; Gaschet, M.; Ploy, M.C.; Perrodin, Y. Ecotoxicity and antibiotic resistance of a mixture of hospital and urban sewage in a wastewater treatment plant. Environ. Sci. Pollut. Res. Int. 2017, 25, 9243–9253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.X.; Zhang, T. Occurrence, abundance, and diversity of tetracycline resistance genes in 15 sewage treatment plants across china and other global locations. Environ. Sci. Technol. 2011, 45, 2598–2604. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.B.; Guo, M.T.; Yang, J. Fate of antibiotic resistant bacteria and genes during wastewater chlorination: Implication for antibiotic resistance control. PLoS ONE 2015, 10, e0119403. [Google Scholar] [CrossRef] [PubMed]
- Calero-Caceres, W.; Muniesa, M. Persistence of naturally occurring antibiotic resistance genes in the bacteria and bacteriophage fractions of wastewater. Water Res. 2016, 95, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Watkinson, A.J.; Murby, E.J.; Costanzo, S.D. Removal of antibiotics in conventional and advanced wastewater treatment: Implications for environmental discharge and wastewater recycling. Water Res. 2007, 41, 4164–4176. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Yuan, K.; Chen, X.; Yang, Y.; Zhang, T.; Wang, Y.; Luan, T.; Zou, S.; Li, X. Metagenomic analysis revealing antibiotic resistance genes (args) and their genetic compartments in the Tibetan environment. Environ. Sci. Technol. 2016, 50, 6670–6679. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, A.C.; Wang, W.; Koteva, K.; Barton, H.A.; McArthur, A.G.; Wright, G.D. A diverse intrinsic antibiotic resistome from a cave bacterium. Nat. Commun. 2016, 7, 13803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Ju, F.; Cai, L.; Zhang, T. Profile and fate of bacterial pathogens in sewage treatment plants revealed by high-throughput metagenomic approach. Environ. Sci. Technol. 2015, 49, 10492–10502. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.J.; Reyes, A.; Wang, B.; Selleck, E.M.; Sommer, M.O.A.; Dantas, G. The shared antibiotic resistome of soil bacteria and human pathogens. Science 2012, 337, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.Z.; Aqil, F.; Ahmad, I.; Ahmad, S. Incidence and transferability of antibiotic resistance in the enteric bacteria isolated from hospital wastewater. Braz. J. Microbiol. 2013, 44, 799–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | p Value | Higher Abundance |
|---|---|---|
| Peptostreptococcaceae | 0.0039 | Downstream to Upstream |
| Afipia | 0.0093 | Downstream to Upstream |
| Holospora | 0.0039 | Downstream to Upstream |
| Azoarcus | 0.0114 | Downstream to Upstream |
| Acinetobacter | 0.013 | Downstream to Upstream |
| Bppunalikevirus | 0.0093 | Downstream to Upstream |
| Yualikevirus | 0.0096 | Downstream to Upstream |
| Sphingobium | 0.01 | Rural to Upstream |
| Kocuria rhizophila | 0.0318 | Residential to Hospital |
| Nitrospira defluvii | 0.0216 | ATE to PCI |
| Caulobacter sp. | 0.0058 | ATE to PCI |
| Afipia clevelandensis | 0.0048 | ATE to PCI |
| Rhodopseudomonas paulustris | 0.012 | ATE to PCI |
| Hyphomicrobium denitrificans | 0.0114 | ATE to PCI |
| Mesorhizobium sp. | 0.0183 | ATE to PCI |
| Paracoccus sp. | 0.0439 | ATE to PCI |
| Reyranella massiliensis | 0.0111 | ATE to PCI |
| Sphingobium xenophagum | 0.0184 | ATE to PCI |
| Sphingopyxis sp. | 0.0003 | ATE to PCI |
| Alicycliphilus sp. | 0.0004 | ATE to PCI |
| Limnohabitans sp. | 0.0005 | ATE to PCI |
| Polaromonas sp. | 0.0003 | ATE to PCI |
| Variovorax sp. | 0.0014 | ATE to PCI |
| Azoarcus sp. | 0.0006 | ATE to PCI |
| Dechloromonas sp. | 0.011 | ATE to PCI |
| Methyloversatilis sp. | 0.0008 | ATE to PCI |
| Thauera aminoaromatica | 0.0212 | ATE to PCI |
| Actinobacter parvas | 0.025 | ATE to PCI |
| Turneriella parva | 0.0058 | ATE to PCI |
| Methanobrevibacter sp. | 0.0357 | ATE to PCI |
| Gordonia amarae | 0.0476 | ATE to PCI |
| Tetrasphera elongata | 0.0218 | ATE to PCI |
| Rhodococcus | 0.0409 | Downstream to FCE |
| Actinobacterium sp. | 0.0116 | Downstream to FCE |
| Polynucleobacter necessarius | 0.00000007 | Downstream to FCE |
| Limnohabitans | 0.00000007 | Downstream to FCE |
| Methylotenera | 0.0404 | Downstream to FCE |
| Bppunalikevirus | 0.0132 | Downstream to FCE |
| Yualikevirus | 0.0266 | Downstream to FCE |
| MetaCyc Pathway | p-Value | Highest Abundance |
|---|---|---|
| PWY-5747 2-methylcitrate cycle II | 0.009 | Upstream |
| PWY-5659 GDP-mannose biosynthesis | 0.010 | Upstream |
| PWY0-42 2-methylcitrate cycle I | 0.010 | Upstream |
| GLYCOGENSYNTH-PWY glycogen biosynthesis I from ADP-d-Glucose | 0.042 | Upstream |
| ILEUSYN-PWY L-isoleucine biosynthesis I from threonine | 0.042 | Upstream |
| PWY-5109 2-methylbutanoate biosynthesis | 0.042 | Downstream |
| PWY-5973 cis-vaccenate biosynthesis | 0.042 | Upstream |
| PWY-6606 guanosine nucleotides degradation II | 0.042 | Upstream |
| PWY-6609 adenine and adenosine salvage III | 0.042 | Upstream |
| PWY-7111 pyruvate fermentation to isobutanol engineered | 0.042 | Upstream |
| PWY-7198 pyrimidine deoxyribonucleotides de novo biosynthesis IV | 0.042 | Upstream |
| PWY-7199 pyrimidine deoxyribonucleosides salvage | 0.042 | Upstream |
| PWY-7208 superpathway of pyrimidine nucleobases salvage | 0.042 | Upstream |
| PWY-7210 pyrimidine deoxyribonucleotides biosynthesis from CTP | 0.042 | Upstream |
| PWY-7211 superpathway of pyrimidine deoxyribonucleotides de novo biosynthesis | 0.042 | Upstream |
| PWY-7663 gondoate biosynthesis anaerobic | 0.042 | Upstream |
| SALVADEHYPOX-PWY adenosine nucleotides degradation II | 0.042 | Upstream |
| UNINTEGRATED | 0.042 | Upstream |
| UNMAPPED | 0.042 | Downstream |
| VALSYN-PWY L-valine biosynthesis | 0.042 | Upstream |
| PWY-3781 aerobic respiration I cytochrome c | 0.042 | Upstream |
| PWY0-1261 anhydromuropeptides recycling | 0.042 | Upstream |
| GLUTORN-PWY L-ornithine biosynthesis | 0.045 | Upstream |
| PWY-6608 guanosine nucleotides degradation III | 0.045 | Upstream |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lambirth, K.; Tsilimigras, M.; Lulla, A.; Johnson, J.; Al-Shaer, A.; Wynblatt, O.; Sypolt, S.; Brouwer, C.; Clinton, S.; Keen, O.; et al. Microbial Community Composition and Antibiotic Resistance Genes within a North Carolina Urban Water System. Water 2018, 10, 1539. https://doi.org/10.3390/w10111539
Lambirth K, Tsilimigras M, Lulla A, Johnson J, Al-Shaer A, Wynblatt O, Sypolt S, Brouwer C, Clinton S, Keen O, et al. Microbial Community Composition and Antibiotic Resistance Genes within a North Carolina Urban Water System. Water. 2018; 10(11):1539. https://doi.org/10.3390/w10111539
Chicago/Turabian StyleLambirth, Kevin, Matthew Tsilimigras, Anju Lulla, James Johnson, Abrar Al-Shaer, Orion Wynblatt, Shannon Sypolt, Cory Brouwer, Sandra Clinton, Olya Keen, and et al. 2018. "Microbial Community Composition and Antibiotic Resistance Genes within a North Carolina Urban Water System" Water 10, no. 11: 1539. https://doi.org/10.3390/w10111539