
Abstract
Inverse electron demand [4+2] Diels–Alder (iEDDA) reactions as well as unprecedented nucleophilic (azaphilic) additions of R-substituted silyl-enol ethers (where R is Phenyl, Methyl, and Hydrogen) to 1,2,4,5-tetrazine (s-tetrazine) catalyzed by \(\hbox {BF}_{3}\) have recently been discovered (Simon et al. in Org Lett 23(7):2426–2430, 2021), where static calculations were employed for calculation of activation energies. In order to have a more realistic dynamic description of these reactions in explicit solution at ambient conditions, in this work we use a semiempirical tight-binding method combined with enhanced sampling techniques to calculate free energy surfaces (FESs) of the iEDDA and azaphilic addition reactions. Relevant products of not only s-tetrazine but also its derivatives such as \(\hbox {BF}_{3}\)-mediated s-tetrazine adducts are investigated. We reconstruct the FESs of the iEDDA and azaphilic addition reactions using metadynamics and blue moon ensemble, and compare the ability of different collective variables (CVs) including bond distances, Social PeRmutation INvarianT (SPRINT) coordinates, and path-CV to describe the reaction pathway. We find that when a bulky Phenyl is used as a substituent at the dienophile the azaphilic addition is preferred over the iEDDA reaction. In addition, we also investigate the effect of \(\hbox {BF}_{3}\) in the diene and steric hindrance in the dienophile on the competition between the iEDDA and azaphilic addition reactions, providing chemical insight for reaction design.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
A growing number of studies about the Diels–Alder (DA) reaction has demonstrated in recent years its prevalence in both traditional and modern organic chemistry, bioinorganic and bioorthogonal chemistry. Among all DA reactions investigated to date, the [4+2] cycloaddition of 1,2,4,5-tetrazines (s-tetrazines) and various dienophiles, referred to as inverse electron demand DA (iEDDA) reaction, is the one that satisfies most bioorthogonal criteria (such as fast, selective, biocompatible and catalyst-free [1, 2]), which are necessary for the use in various applications ranging from protein labeling to cancer imaging or materials science [3].
It has been well known that the DA reaction generally occurs as a major outcome when s-tetrazine reacts with common dienophiles. However, our recent work in close collaboration with the Gademann group reported an unprecedented nucleophilic addition reaction of 3-bromosubstituted s-tetrazine (3-Br-s-tetrazine) which is in contrast to [4+2] cycloaddition reactions [4]. Moreover, the direct nucleophilic attack to nitrogen in s-tetrazine has rarely been described in the literature and is generally restricted to 1,2,4,5-tetrazine and other tetrazine derivatives such as 1,2,3,4-tetrazine and 1,2,3,5-tetrazine [5,6,7]. The investigation performed by our experimental collaborators revealed a surprising discovery in reactivity of 3-monosubstituted s-tetrazines, a small s-tetrazine building block, with silyl-enol ethers for the synthesis of e.g. macromolecules [4]. In this work, we, therefore, focus on chemical competition between [4+2] cycloadditions and azaphilic addition reactions of 3-Br-s-tetrazine and silyl-enol ethers. In addition, one main challenge is to observe the aforementioned reactions and their dynamics at a molecular level in the experiments since, e.g. most of the single-step reactions occur very fast which makes it difficult to catch up with the desired products. To address this issue, computational simulations can be used to provide insight by mimicking the chemistry and allowing additional information about processes at the molecular level behind the iEDDA and azaphilic additions of s-tetrazines, providing meaningful details not detectable with only experimental techniques.
Usually the polar effect of the substituent and the stabilization of valence electrons in the s-tetrazine control the type of DA reaction. It has been recognized that s-tetrazines are good candidates as dienes for the iEDDA reaction. Moreover, a Lewis acid can be used as a catalyst to activate the s-tetrazine by lowering the level of its lowest unoccupied molecular orbital (LUMO), for which corresponding iEDDA reactions with electron-rich dienophiles could occur under relatively mild conditions and arrive at the desired product in the form of a bridged structure [8, 9]. A number of Lewis acids, e.g. \(\hbox {SO}_{3}\), \(\hbox {Br}_{2}\), and \(\hbox {AlCl}_{3}\), have been used. In particular, boron halides possess low-lying orbitals that are suitable for the cycloaddition reaction. The electrophilicity of boron in Lewis acids increases in the order of \(\hbox {BBr}_{3}\) \(\rightarrow\) \(\hbox {BCl}_{3}\) \(\rightarrow\) \(\hbox {BF}_{3}\). Hence it has been proved by several calculations [10,11,12] and experiments [13, 14] that \(\hbox {BF}_{3}\) is a highly efficient catalyst for the iEDDA reactions [1]. At the same time, based on experimental results reported in the previous study, the reaction could produce the product with up to 82% yield in the presence of \(\hbox {BF}_{3}\) [4]. In addition, the \(\hbox {BF}_{3}\) Lewis acid bound to the diene can indirectly influence its electronic state via electron delocalization through the N-containing aromatic ring [15]. Therefore, it has also been important to investigate the effect of \(\hbox {BF}_{3}\) on chemical selectivity of which different positions of nitrogen atoms on the s-tetrazine ring are compared [16, 17].
From a computational point of view, models have been developed over decades to mimic chemical reactions and capture physical/chemical changes at the atomic level. Various reactions were used as example tests for the developed method’s ability evaluation. The DA reaction is one of the popular choices as it can take place via concerted or stepwise mechanisms, depending on the experimental conditions. Both static Density functional theory (DFT) and (classical) molecular dynamics (MD) calculations were performed to investigate the dynamically concerted mechanism of the DA reaction [18,19,20,21,22]. For the unusual regio- and stereo-selectivity of DA reactions, selected works related to the inverse DA reaction are mentioned in the following. Shrivastav et al. studied the reaction energetics and kinetics of 2-pyrone complex undergoing the retro DA reaction [23]. They also calculated the free energy surface (FES) of the DA reaction using Car-Parrinello molecular dynamics with MetaD to obtain the Gibbs activation energy. Li et al. used quantum mechanics/molecular mechanics and Multistate Bennett Acceptance Ratio and Weighted Thermodynamic Perturbation methods to calculate the FES of a similar DA reaction [24]. These notable studies warrant that the FES is a crucial descriptor to shed light on theoretical explanation of the DA reaction.
A growing number of significant developments of simulation techniques to calculate the FES have been presented, in particular enhanced sampling techniques, which have been integral to many discoveries in chemistry and provide attractive tools including umbrella sampling [25], MetaD [26,27,28] and Blue Moon (BM) ensemble [29]. Previous studies addressed by the MetaD method have proven a wide range of applications in chemical reactions which can be described by reaction coordinates or the so-called collective variables (CVs). The CVs have to be chosen in such a way that various conformations can be distinguished and the validity of the reaction pathways can be demonstrated by the FES.
The aim of this work is to characterize the kinetics and the thermodynamics behind the iEDDA and azaphilic additions of 3-Br-s-tetrazine (with and without \(\hbox {BF}_{3}\)) and silyl-enol ethers (as dienophile) with R substituent group; where R is Phenyl (Ph), Methyl (Me), and Hydrogen (H) (see Fig. 1), providing unprecedented free-energy profiles by means of enhanced sampling-based MetaD and BM ensemble simulations. DFT-based molecular dynamics (DFT-MD) has been employed for a wide range of applications ranging from biological systems, over material design, to molecular chemical modeling [30,31,32,33,34]. However, DFT-MD can become computationally demanding, depending, among others, on the size of the modeled system and process to be studied. In order to speed up our simulations, we adopt a tight-binding (TB) method [35, 36], namely Geometry, Frequency, Noncovalent, eXtended TB (GFN-xTB) based on already defined semi-empirical parameters which allow to reduce the computational time required (in comparison with standard DFT-MD algorithms). The use of GFN-xTB has been gradually growing with a wide range of applications for thermodynamics calculations [37, 38], transition metal complexes [39, 40], and spectroscopy [41,42,43,44]. The aim of this work is to complement our previous studies by the inclusion of finite temperature effects, explicit solvent, and the effect of substituents on the dienophile and to provide additional computational insight into the studied systems as well as the role of \(\hbox {BF}_{3}\).
In the following sections, we first give an overview of WT-MetaD and BM methods that we used for performing the density functional tight binding MD (DFTB-MD) simulations. A clear and meaningful discussion on the selection of CVs for WT-MetaD and BM is also accommodated in the study of the FES. In this work, not only standard CVs are employed, but we also utilize recently developed adjacent-contact-matrix-based atomic coordinate functions including Social PeRmutation INvarianT (SPRINT) coordinates and path nuclear collective variable (path-CV), which have been successfully adopted in Ref. [45] on the formation of fullerene from graphene and in Ref. [46] on methanol oxidation.

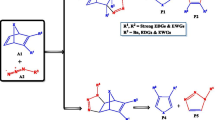
Inverse electron demand Diels–Alder (iEDDA) and nucleophilic (azaphilic) addition reactions. A schematic reaction in the box shows the \(\hbox {BF}_{3}\)-mediated s-tetrazine adduct. \(\hbox {BF}_{3}\)@N\(_X\) denotes binding of \(\hbox {BF}_{3}\) at the X position of nitrogen atom in the ring
The remainder of the paper proceeds with the structural analysis in order to assess the conformation change of reaction partners along WT-MetaD simulations. We focus on azaphilic addition reactions and show that the 3-Br-s-tetrazines are stereoselective to the replacement of substituent (R group) in the silyl-enol ether. Moreover, the direct attack of \(\hbox {BF}_{3}\) to different positions of nitrogen in 3-Br-s-tetrazine plays an important role in the energetics of the reaction, leading to chemical selection between normal iEDDA and nucleophilic addition reactions.
2 Methods
2.1 Computational Details
We use a cubic box with a side length of 15 \({\text{\AA} }\) size containing one DA product and 31 dichloromethane (DCM) molecules in order to mimic the experimental density of 1.33 \(g/cm^3\). Periodic boundary conditions (PBCs) are applied in all three spatial directions. The box size and number of molecules were chosen as the best compromise between the computational complexity of the system and the reliability of the calculations in reproducing the expected physical/chemical properties. For the DFTB-MD simulations, the GFN-xTB [35] method based on semiempirical parameters in the CP2K package version 7.1 [47] was used. The GFN-xTB basis set was employed with an auxiliary plane-wave basis set using a cutoff of 800 Ry. For the DFT-MD simulation, the atoms were instead described by the DZVP-MOLOPT-SR-GTH basis set and GTH-PBE pseudopotentials [48,49,50]. The Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional [51] and Grimme’s D3 dispersion correction [52] were employed.
All systems containing the product of either iEDDA or azaphilic addition reaction were equilibrated using isothermal-isobaric (NPT) followed by canonical (NVT) ensembles with Nose-Hoover thermostat using a time constant of 100 fs using an MD time step of 0.5 fs at a constant temperature of around 300 K. We note here that, for NPT ensemble, in addition to the thermostat, a barostat is used with the pressure set to 1 Bar and a time constant of 80 fs. Unbiased MD simulations were carried out for 25 ps, whereas biased simulations were carried out for 50 ps. PLUMED version 2.6 [53] was used as a plugin in conjunction with QUICKSTEP [54] in CP2K for driving WT-MetaD simulations and reconstructing the FES. Helmholtz free energies of iEDDA and azaphilic addition reactions were computed.
All static calculations were carried out by means of DFT using TURBOMOLE version 7.3 [55]. For DFT calculations, we adopted computational parameters tested and used in the previous work [4]. The structures were optimized using BP86 [56, 57] with a def2-TZVP [58] basis set. Single-point (SP) energies were computed using B3LYP [59] at the same level of basis set. In addition, geometry optimization and SP energy calculations were also performed with PBE functional and def2-SVP basis set. Grimme’s 3D dispersion correction was employed. The solvent effects of DCM were considered using the COSMO model [60] (dielectric constant = 8.93) in TURBOMOLE. Vibrational frequency calculations were carried out to verify the TS and obtain Gibbs free energies.
2.2 Metadynamics
MetaD is developed based on a history-dependent Gaussian bias potential using the so-called CV (\(\xi\)) which usually relies on nuclear coordinates. For the sake of brevity, we do not explicitly note here the dependence of \(\xi\) on e.g. nuclear coordinates. Within a standard MetaD method, the sum of Gaussian potentials deposited along the reaction coordinate and simulation time is given as
where \(\xi _i\) is the value of the i-th CV at time t, \(\xi _i(t')\) at time \(t'\), d is the number of CVs, W the height, \(\tau\) the deposition stride, and \(\sigma _i\) the width of the Gaussian potential used for the i-th CV. In standard MetaD, Gaussian bias potential with constant W is accumulated during the entire simulation. This might lead to overfilling of the configurational space.
To avoid this shortcoming, well-tempered metadynamics (WT-MetaD) developed by Barducci et al. has been introduced with a slow-converged potential at the final state of the calculation [61]. The height of the Gaussian potential is controlled with decreasing simulation time
where \(W_0\) is the initial height of the Gaussians potential and \(\varDelta T\) is a parameter specifying rescaling rate with the dimension of temperature. To perform WT-MetaD simulations with the PLUMED package, the so-called “bias factor” \(\gamma\) is chosen which is defined as follows:
where \(T + \varDelta T\) is the temperature of the CVs and T is the system temperature. When choosing \(\gamma\) care has to be taken to ensure that the free energy barriers can be efficiently crossed in the appropriate simulation time scale [53].
Once the metadynamics simulation has converged at a longer time (\(t\rightarrow \infty\)), free energy F as a function of \(\xi\) can then be analytically computed by integrating Eq. 1,
2.3 Blue Moon Ensemble
In the BM ensemble, the FES is calculated as a function of the degree of freedom of a given reaction coordinate \(\xi ' \equiv \xi '(\{r_{}\})\), which in general depends on a subset of N nuclear positions \(\mathbf{r}_{\mathrm{i}}\). Integration of average forces \(F_{\xi^{\prime}}\) results in the free energy profile given by
The average force \(F_{\xi^{\prime}}\) is derived from the Lagrange multiplier \(\lambda\) according to
for which \(k_{\text {B}}\) is the Boltzmann constant. The remaining terms, Z and G, are the so-called “Fixman mass-metric tensor correction” and correction factors associated with the transformation from generalized constraints to Cartesian coordinates, respectively. They are defined as
and
where \(m_i\) is the atomic mass of nucleus i, and the corresponding position vector is \(\mathbf {r}_{\varvec{i}}\)[62]. When the bond distance \(r_{ij}\) is chosen as CV, both Z and G become constant values [63]. Therefore, Eq. 6 can be rewritten as
In all other cases where the order parameter involves multiple distances, Z and G have to be derived analytically and evaluated for each step of the simulation.
3 Results and Discussion
3.1 Studied System
We studied the DA reactions of 3-Br-s-tetrazine and silyl-enol ether with R substituents (where R is Phenyl, Methyl, or Hydrogen) in DCM solvent. Systematic evaluation of the system in equilibrium resulting in the appropriate steady-state size of a cubic simulation box with side length of 15 \({\text{\AA} }\) side has been performed using DFTB-MD with the GFN-xTB method (see Fig. 2). We tested our computational setup calculating common chemical and physical parameters of the pure DCM system for which the data are reported in Table 1. Those primary data include density \(\rho\), enthalpy of vaporization \(H_{vap}\), and viscosity \(\eta\). More information regarding \(\varDelta H_{vap}\) and \(\eta\) can be found in the Supporting Information (SI). We note that the energy related to the association of the two reaction partners and corresponding reorganization of the solvent is not considered in this work.

The periodic simulation box containing silyl-enol ether with Phenyl substituent group, 3-Br-s-tetrazine, and 31 DCM molecules
Further calculations of radial distribution functions (g(r)) of pure DCM were carried out and they are available in the SI. The g(r) by GFN-xTB indicates that the configuration distribution of molecules for the C–C pair has a peak around 4.72 \({\text{\AA} }\), while the one of the Cl–Cl pair is around 3.05 \({\text{\AA} }\). These findings are in good agreement with those by DFT-MD: 4.85 \({\text{\AA} }\) for the C–C pair and 3.39 \({\text{\AA} }\) for the Cl–Cl pair, and experimental g(r) of 4.80 \({\text{\AA} }\) for the C–C pair of the same system.
3.2 Choosing Collective Variables
We use the standard WT-MetaD for one-dimensional and two-dimensional FES simulations (parallel-bias WT-MetaD [67]) to enhance the crossing of high energy barriers for iEDDA and azaphilic addition reactions. Choosing CVs matters the simulation as they characterize the occurrence of long-lived metastable states [68]. That is, CVs have to be defined as a physical change parameter that drives the reaction coordinates or region where the reaction takes place. Moreover, the identification of appropriate set of CVs is largely dependent on and specific to the system.

Outlines of a DA and b azaphilic additions with the different CVs sets. a \(D_{1}\) and \(D_{2}\) refer to specific \(C_{1} \cdots C_{3}\) and \(C_{2} \cdots C_{4}\) bond distances of the reaction (used as CVs), respectively. \(S_{i}\) coordinates are computed and sorted from chosen four carbon atoms: \(C_{1}\), \(C_{2}\), \(C_{3}\), and \(C_{4}\), resulting in \(S_{1}\), \(S_{2}\), \(S_{3}\), and \(S_{4}\). (b) \(D_{i}\) refers to specific bond distance between \(C_{2}\) and i-th N atom. We note here that for the C-N bond distance of the azaphilic addition there are only two SPRINT coordinates used. Other atoms are not shown for the sake of simplicity
In our case, both iEDDA and azaphilic additions are characterized by concerted mechanisms in which bond forming and bond breaking take place in a single step [8], therefore the most straightforward way is to use the bond distance between 3-Br-s-tetrazine and silyl-enol ether as CV. As shown in Fig. 3, bond distances (\(d_{1}\) and \(d_{2}\)) and the sum of the two bond distances (\(D = d_{1} + d_{2}\)) are straightforward CVs to represent the (time) evolution of the reaction, and are used to perform two- and one-dimensional WT-MetaD simulations, respectively. We also use the SPRINT coordinate [69] given by
with
\(\lambda ^{max}\) and \(\varvec{\nu }^{max}\) are respectively the maximum modulus eigenvalue and the corresponding eigenvector of adjacency matrix (\(\varvec{A}\)) with relation values between two atoms i and j of the system containing N atoms. Superscript sorted indicates that \(\varvec{\nu }^{max}\) is sorted in ascending order where identical atom types are combined together into one value. \(a^{M}_{ij}\) is the number of walks of length M connecting atoms i and j [69]. In this work we set M to be 1 [69]. The \(a_{ij}\) element of \(\varvec{A}\) is defined by the following switching function
where \(r_{ij}\) and \(r_{0}\) are the distance from atom i to atom j and its corresponding bond cutoff for the specific pair of atoms used to evaluate \(r_{ij}\) with the strength parameters n and m, which is usually chosen based on the typical type of bond, in this work set to be 6 and 12 which were adopted from previous work [70]. The values for \(r_{0}\) were set to be 2.65 \({\text{\AA} }\) for the C-C bond and 2.22 \({\text{\AA} }\) for other atomic interactions determined from distribution functions [70].

Path \(S(\mathbf{R} )\) connecting reactant state A and product state B, and distance \(Z(\mathbf{R} )\) from the reference path \(S(\mathbf{R} )\) to metastable state C
The path-CV developed by Branduardi et al. [71] is another possible choice for a CV. The path-CV has been used to study complex chemical systems and calculate their associated FESs [72,73,74], allowing one to compute the progress along the high-dimensional reaction path \(\varvec{R}\) and the distance \(Z(\mathbf{R} )\) from the (high-dimensional) reference path (see Fig. 4). We refer the interested readers to Ref. [75] for more details.
In this work, matrices of reactant state A and product state B are used as initial and final frames in path-CV to generate the reference path \(\varvec{R}\) for describing the evolution of the azaphilic addition. Contact matrices—adjacency matrices in which two atoms are adjacent if they are bonded—used for constructing path-CV of the iEDDA and azaphilic addition reactions are provided in the SI.
3.3 Free Energy Surface Calculations
3.3.1 Metadynamics Simulation
WT-MetaD offers ways to use different and multiple CVs to calculate the FES of a given reaction. We adopt the WT-MetaD technique to treat iEDDA and azaphilic addition reactions performing WT-MetaD simulations with a number of possible combinations of the WT-MetaD parameters related to the Gaussian hills bias potential. For example, we varied the width and the height of the potential that the WT-MetaD employs to force the system away from the trap and travel to unexplored regions in the phase space. Table S1 in the SI shows a list of the potential parameters used, including deposited Gaussian potential, frequency or rate of hill deposition (pace), and upper and lower walls of the CV which are values that limit the region of the phase space (a function of CV) accessible during the sampling. In addition, we have checked whether the upper or lower walls have influenced our WT-MetaD calculations, and all the free energy landscapes calculated in our work are not affected by the upper or lower walls employed, providing therefore reliable reaction pathways and related energies.

Time-progress FES of the first occurrence of iEDDA reaction in DCM solution for Ph derivative using WT-MetaD and C–C bond distances (CV1 and CV2) as CVs. The phase space starts with the product state and proceeds to the reactant state. Frame order: from left to right, top to bottom. The time difference between plots is 0.5 ps
iEDDA Reaction An important visualization of the evolution of the enhanced sampling simulation of the iEDDA reaction for the Ph derivative can be seen in Fig. 5. The time-progress profile of the FES shows a continuous biased energy surface and points out the existence of the product and reactant, respectively, as the former was the starting state. As can be seen from the first few snapshots, the system was activated by filling potentials and the process continued to the reactant after the simulation has been done for 2 ps. After that, the obtained FES evolution proceeds until the phase space is stationary and two metastable states were found within a defined region restricted by the wall parameter in the WT-MetaD simulation setup. Moreover, convergence is achieved when the simulation freely moves between the states of interest. This is equivalent to the reoccurring observation of the chemical transformation of interest, showing multiple back-and-forth events which refer to the occurrence of the desired iEDDA reaction. We deliberately went beyond the simplification in this study in order to get a complete perspective of both the reactant and product states for the relevant reactions, as well as their free energy differences in comparison with experimental observes.

FESs of iEDDA reaction of a Ph, b Me, and c H derivatives using WT-MetaD and the two bond distances as CVs. A white dash line indicates the MEP passing through local minima and TS

Selected reactant, TS, and product structures of iEDDA addition for Ph derivative in DCM solution obtained by metadynamics using the two bond distances as CVs. Bond distances are in \({\text{\AA} }\). The red arrows denote the vibrational motion observed around the TS. The snapshots were generated from the production phase of converged WT-MetaD simulation
The energy landscape using \(\hbox {C}_1-\hbox {C}_3\) and \(\hbox {C}_2-\hbox {C}_4\) distances as CVs in Fig. 6 clearly shows two separate local minima of reactant and product states of the [4+2] iEDDA reaction on 3-Br-s-tetrazine, and minimum energy path (MEP) calculated using MEPS [76]. Structures of reactant, TS, and product corresponding to states of the FES taken from the WT-MetaD trajectory are shown in Fig. 7. The activation energies (free energy barrier between reactant and TS) obtained from the first occurrence of the reaction from WT-MetaD using bond distances as CVs for Ph, Me, and H derivatives (without \(\hbox {BF}_{3}\)) are about 26.72 kcal/mol, 25.83 kcal/mol, and 18.76 kcal/mol, respectively. In addition, SPRINT and path-CV give the activation energies of the iEDDA reaction for Ph derivative of 27.22 kcal/mol and 33.36 kcal/mol, respectively, clearly showing that both of these CVs suggest that the iEDDA reaction is unfavorable when the phenyl is used as a substituent. However, when \(\hbox {BF}_{3}\) adduct is used, the SPRINT coordinates indicate a contrary preference of azaphilic addition (20.02 kcal/mol) over the iEDDA reaction (24.94 kcal/mol) for Ph derivative in agreement with experimental findings [4]. The energetic preferences of azaphilic addition and iEDDA reactions (however without \(\hbox {BF}_{3}\)) using SPRINT coordinates are as follows: 28.28 vs 24.28 kcal/mol for Me and 22.80 vs 22.01 kcal/mol for H. Those reported values are in very good agreement with the values obtained by means of static DFT calculations (see Table 2). We note that the \(\hbox {BF}_{3}\) adducts of Me and H derivatives were not considered in this work, which hampers direct comparison to experiment, but we refer the reader to Ref. [4] for more details on experimental observation and associated static DFT calculations. Another remark also shows that the SPRINT coordinates can estimate free energies of the iEDDA reaction that are in good quantitative agreement with the DFT-2 setting for static DFT calculation.

FESs of iEDDA reaction for Ph derivative in DCM solution obtained using WT-MetaD and different SPRINT coordinate sets as CVs: a \(S_{1}\) and \(S_{2}\), b \(S_{1}\) and \(S_{3}\), c \(S_{1}\) and \(S_{4}\), d \(S_{2}\) and \(S_{3}\), e \(S_{2}\) and \(S_{4}\), and f \(S_{3}\) and \(S_{4}\). A white dash line indicates the MEP passing through local minima and TS
Figure 8 shows FESs as a function of different SPRINT coordinates chosen from four carbon atoms (in both diene and dienophile) involving bond breaking in the iEDDA reaction. A list of pairs of the SPRINT coordinates is shown in Table 3. Each pair is selected based on the importance of the SPRINT coordinate that is defined as the actual value of SPRINT sorted from the highest coordinate (\(S_1\)) to the lowest coordinate (\(S_4\)). According to the property of SPRINT, each SPRINT coordinate can carry global information of the topology of the network and includes local information of surrounding atoms to the SPRINT coordinate of interest. As shown in the figure, only the first two FESs using \(S_1\)/\(S_2\) (see 8a) and \(S_1\)/\(S_3\) (see 8b) can reasonably reconstruct the energy pathway for iEDDA from product to reaction (reversely), especially, in the case of \(S_1\)/\(S_2\) the metastable states can be distinguished. On the other hand, the reconstruction ability of WT-MetaD to reach the desired FES decreases when the low-value SPRINT coordinates, such as \(S_2\)/\(S_4\) (see 8e) and \(S_3\)/\(S_4\) (see 8f), are used as biased CVs, losing the possibility to find local minima of the state of interest. This also shows the impact of reordering the SPRINT coordinates that are associated with different atoms. It is worth mentioning that in the cases where the simulation is unable to quantify the desired metastable states and when the free energy difference between the reactant and product states is not of interest, it is common to stop MetaD simulations after an unexpected reaction is found.
A few comments can be added on the relationship between the evolution of the CVs and structural change of the TS of the iEDDA reaction. As can be seen in Fig. 6, all FESs obtained with C-C distances as CVs show that the iEDDA reactions using Ph, Me, and H derivatives share the same reaction pathway. As the simulation started in in the product state with stationary \(C_{1}{\cdots }C_{3}\) and \(C_{2}{\cdots }C_{4}\) distances of 1.60 \({\text{\AA} }\), the system was then forced to travel to the nearby region along the CV1 (\(C_{1}{\cdots }C_{3}\)) rather than CV2 (\(C_{2}{\cdots }C_{4}\)), from about 1.60 \({\text{\AA} }\) to 3.20 \({\text{\AA} }\) for CV1. The simulation then proceeded to the TS with a slight increase of CV2 from about 1.75 \({\text{\AA} }\) to 2.09 \({\text{\AA} }\). In contrast, for the route from the TS to reactant state, the system was instead mainly governed by the \(C_{2}{\cdots }C_{4}\) distance with a drastic change of CV2 from 2.09 to 3.95 \({\text{\AA} }\) and of CV1 from 3.20 to 4.00 \({\text{\AA} }\). The overall evolution of the reaction from product to reactant states and vice versa corresponds to the oscillation of \(C_{1}{\cdots }C_{3}\) and \(C_{2}{\cdots }C_{4}\) bonds. When comparing with the FESs using \(S_1\)/\(S_2\) and \(S_1\)/\(S_3\) CVs, the pathways of the iEDDA reaction shown in Fig. 8a and b manifest the direct route via the TS which is located in a narrow region. Both of these two SPRINT coordinates’ FESs are symmetric and show identical characteristic of the stable states compared to other SPRINT coordinates’ FESs such as \(S_1\)/\(S_4\) since the SPRINT coordinates start from around 0.8 to 2.6 (arbitrary unit) in both directions (\(S_{1}\)/\(S_{2}\) and \(S_{1}\)/\(S_{3}\)). Another interesting difference between these two FESs is the location of the TS. In the case of \(S_{1}\)/\(S_{2}\), the TS was found to be in between the reactant and product states, indicating the balance between the \(S_{1}\) and \(S_{2}\) coordinates that they are almost equal to each other in terms of the importance of the components in SPRINT coordinates, while in the other case where \(S_{3}\) was used instead the TS was shifted to the lower \(S_{1}\) and \(S_{3}\) components, corresponding to the imbalance between \(S_{1}\) and the lower-important component, i.e., \(S_{3}\) or \(S_{4}\).

FES of the azaphilic addition in DCM solution (no \(\hbox {BF}_{3}\) adduct) using WT-MetaD and C-N distance (Ph, Me, and H). C-N distances (\({\text{\AA} }\)) for each structure are as follows: A 3.40, B 2.81, C 2.42, D 2.22, E 1.90, and F: 1.50

FESs of the azaphilic addition for Ph in DCM solution using WT-MetaD and different CVs: a SPRINT coordinates and b path-CV. A white dash line indicates the MEP passing through local minima and TS

Selected reactant, TS, and product structures of azaphilic addition for Ph derivative in DCM solution obtained by metadynamics using the two bond distances as CVs. Bond distances are in \({\text{\AA} }\). The red arrows denote the vibrational motion observed around the TS. The snapshots were generated from the production phase of converged WT-MetaD simulation
Azaphilic Addition Reaction When Me substituted silyl-enol ether reacted with 3-Br-s-tetrazine, the pyridazine was observed as the minor product of this transformation in low conversion together with a major unknown compound [1]. When bulky partner Ph substituted silyl-enol ether has been used in the azaphilic addition, in this work, we found that the reaction changes from cycloaddition to azaphilic addition when using SPRINT coordinate as CVs and \(\hbox {BF}_{3}\), mirroring the previous work which found that the latter is an energetically favored reaction rather than the former [4]. This is in line with the experimental finding in the previous work that the ability of the dienophiles for the azaphilic addition depends strongly on the steric demand of the alkyl and aryl substitutes [4].
The FESs of the azaphilic addition reaction obtained with the C-N distance as CV are shown in Fig. 9 with structures along the reaction pathway. The FESs obtained using SPRINT coordinates or path-CV as CVs are shown in Fig. 10. The MEP also shows the direct route from reactant to product states via the TS. In the particular case of the unexpected azaphilic addition reaction of the Ph derivative, the activation energies obtained from the FES using WT-MetaD simulation are about 37.62 and 32.79 kcal/mol for using the C-N bond distance and their SPRINT coordinates, respectively, as CVs. When the \(\hbox {BF}_{3}\) catalyst was used, the activation energies for adducts with the \(\hbox {BF}_{3}\) attached at N-1 and N-2 positions of the ring gradually decrease to 28.90 and 20.58 kcal/mol, respectively, for bond distance CV, and to 25.37 and 20.02 kcal/mol, respectively, for the SPRINT CVs. These activation energies are consistent with the static DFT’s energy barriers measured as free energy differences between the optimized geometry of the associated reactant and the TS. However, we also noticed that the dynamic simulations using the SPRINT CVs overall yield lower activation energies than using bond distance CV or path-CV. The difference of this estimated activation energy between using the SPRINT and other CVs might be attributed to the slightly different TS structures obtained, and/or localization of selected atoms in a molecule, which could directly affect the lower eigenvalue of the corresponding eigenvector of the adjacency matrix used to calculate the SPRINT. We also notice that path-CV gives the activation energy of about 24.62 kcal/mol for the H derivative azaphilic addition, whereas, the C-N distance gives the lowest free energy of 13.95 kcal/mol among other methods in Table 4. A large difference between the activation energies using the C-N distance and the path-CV (10.67 kcal/mol at most in the case of H derivative) indicates that the transformation of CV from internal coordinate to geometric coordinate could manufacture different characteristics of the FES of the same reaction.
In our previous study on the investigation of the electrophilicity of each backbone atom in 3-Br-s-tetrazine for the same kind of reaction, we have found that the abovementioned steric effect helps to rationalize the reactivity of 3-Br-s-tetrazine towards azaphilic addition against iEDDA reaction [4]. The most electrophilic site was identified as a nitrogen atom that is spatially opposite to the \(\hbox {BF}_{3}\) attachment and found to undergo the azaphilic addition reaction. Moreover, the results revealed that nucleophilic attack on aromatic compounds without elimination is not favored, as aromaticity is not restored. Moreover, committor analysis was performed to describe the ability of bond distance as CVs in WT-MetaD simulations (see the SI for more details). The analysis clearly showed that the bond distances are reasonable to capture the transition between reactant and product for the iEDDA and azaphilic addition reactions.
Structural Analysis To gain additional insight into the evolution of both iEDDA and azaphilic addition reactions, further analysis is desirable for providing a quantitative estimation of the reaction progress. Root mean square deviation (RMSD) plots are general tools that can be used to track the structural change of molecules in the simulation and to measure the quality of reproduction of the reaction. RMSD values depend on the interaction between reaction partners. We computed the RMSD of atomic positions of selected studied derivatives using the trajectory from the biasing simulation (see the SI for all RMSD plots). Generally, a low value of individual RMSD reflects the high significant pose of the structure of interest onto the reference structure. Selected snapshots for nuclear structures of the reactant, TS, and product states obtained from the first occurrence of the reaction in WT-MetaD trajectory were used as the reference structures (see Figs. 7 and 11).

Partial g(r) of DCM solution with Ph derivative in the reactant state for C–C, Cl–Cl and H–H distances at 300 K and 1 Bar under PBC. The g(r) was computed with a spherical shell of thickness \(\delta r\) of 0.01 \({\text{\AA} }\)

Partial g(r) of DCM solution with Ph derivative in the reactant state for C–Cl and H–Cl distances at 300 K and 1 Bar under PBC. The g(r) was computed with a spherical shell of thickness \(\delta r\) of 0.01 \({\text{\AA} }\)
Moreover, we computed the radial distribution functions g(r) for the Ph derivative to investigate the influence of the DCM solvent. In principle, the first peak of the computed g(r) represents the probability to find DCM in the corresponding first solvent layer (see Figs. 12 and 13). At the short distance from the reference point, the bulk DCM is calculated to be at around 1.5 \({\text{\AA} }\) to 3.0 \({\text{\AA} }\). We also found that the computed g(r) of DCM is closely related to the RMSD of the solute using the product state as the reference structure for both the iEDDA and azaphilic addition reactions at the initial biasing simulation for 1.9 ps. That is, the solute and DCM molecules were consolidated and remained in the structure of the product state which is the most stable state compared to other states in the trajectory. Moreover, during the course of the enhanced sampling simulation, DCM molecules undergo structural reorganization which then affects the stability of the structure of the solutes as we found a gradual increase of the rate of its RMSD movement (see the SI for more details). This finding is a piece of evidence that the explicit solvent in the enhanced sampling simulations plays a non-negligible role for the iEDDA and azaphilic addition reactions, which is not necessarily captured well in the static calculation where the implicit solvent continuum model was used.
3.3.2 Blue Moon Ensemble Simulation

Free energy pathway of the iEDDA reaction of 3-Br-s-tetrazine for Ph derivative in DCM solution using BM ensemble simulation
In addition to the WT-MetaD method, the energy path quantified by the BM ensemble can provide the energy profile of the reaction. The BM ensemble is run in a similar way to that of the MetaD. We estimate the Lagrangian of the system at different constraints. Since all BM simulations of Ph, Me, and H share the computational workflow and analysis, the case of Ph is selected as representative for results and discussion. The Lagrangian (constraint forces) and free energy obtained from the BM simulations for Ph as a function of bond length are shown in Fig. 14 for \(d(C_{1} \cdots C_{3}) + d(C_{2} \cdots C_{4})\). The structure of reactant, TS, and product corresponding to the local minima and maxima are consistent with those obtained by the MetaD, confirming the consistency in terms of phase space. The corresponding activation energies by means of BM ensemble for iEDDA and azaphilic additions of Ph, Me, and H are included in Tables 2 and 4, respectively.
A direct comparison of activation energies clearly indicates the feasibility of the two different reaction pathways in good agreement with static DFT calculation and the WT-MetaD simulations in terms of qualitative tendency, in ascending order of the size of substituent: H, to Me, and Ph. The overall free energies of the iEDDA reaction of substrates without \(\hbox {BF}_{3}\) decrease from 20.48 to 17.18, and to 12.57 kcal/mol, respectively. When the iEDDA reaction takes place in the presence of the \(\hbox {BF}_{3}\) catalyst, the energy barrier is reduced. However, in the case of azaphilic addition, all free energies are still high compared to the iEDDA reaction. For example, BM simulation of azaphilic addition using H as substituent gives free energy of 16.53 kcal/mol, which is close to that of 17.18 kcal/mol for the iEDDA reaction using Me as substituent. In contrast to WT-MetaD simulations, the BM method predicts in most cases the activation energies less quantitatively accurate compared to the values obtained by all other CVs utilized in the WT-MetaD, showing the weakness of this constraint-based ensemble method [63]. In addition, underestimation of the energy barrier on the FES computed by BM ensemble is noted which might be due to the fact that the bond distance used as constraint does not capture the exact transition state (TS) structure. Therefore, the qualitative accuracy in the calculation of the free energy by BM ensemble depends on the resolution and the degree of freedom of the constraint used in the simulation.
4 Conclusion
In this work, we computationally investigate the chemical reactivity of inverse electron demand Diels–Alder (iEDDA) and nucleophilic (azaphilic) addition reactions between 3-Br-s-tetrazine (with or without \(\hbox {BF}_{3}\) adduct) and alkyl substituted silyl-enol ethers in dichloromethane (DCM) solvent. The effect of Phenyl (Ph), Methyl (Me), and Hydrogen (H) substituents on dienophile is also systematically investigated. Enhanced sampling-based metadynamics (MetaD) and blue moon (BM) ensemble were used to reconstruct free energy surfaces (FESs) and calculate activation energies. The dynamic calculations can provide a comprehensive perspective of the studied reaction in terms of FESs with the description of explicit DCM solvent at ambient conditions, whereas in static DFT calculation explicit solvent can cause unreliability of the estimation of activation energy [77, 78].
We adopted the Well-Tempered MetaD (WT-MetaD) that can help where standard MetaD lacks the convergence in the reconstruction of FESs in a single simulation run. The convergence of the simulation is analyzed in order to characterize the multidimensional free energy landscape of the system and to understand rare events. A direct comparison between energetic differences of the reaction pathway simulated by either WT-MetaD method, BM ensemble, or static DFT model is mirroring in terms of quantitative accuracy with respect to the different substituents (Ph, Me, and H) and the trend of reaction feasibility. There is a remarkable agreement among the nuclear structures of the reactant, transition state, and product. We also found that the steric hindrance in the ligand of dienophile plays an important role in the selectivity of the reaction.
In comparison to previous static DFT calculations with implicit DCM solvent, structural analysis on the configuration of explicit DCM molecules in WT-MetaD shows that dynamic reorganization of DCM can stabilize the interaction between solute and solvent. The RMSDs with different reference states were also computed to study the transformation of the iEDDA and azaphilic molecules via the reaction pathway. The crossing of RMSDs between reactant and product references shows that the reaction does not take place immediately but is delayed after (small) rearrangement of DCM solvent molecules [79].
There is a promising chance to explore the ambiguous chemistry of s-tetrazines and silyl-enol ethers using other collective variables (CVs) even though those were developed for specific problems. We gain a much better understanding of the importance of structural descriptors from dynamic simulations, especially helping one carefully choosing the appropriate CVs for similar DA and azaphilic addition reactions or other related chemical reactions. Our computational WT-MetaD results reveal that internal-based CVs used in this work, SPRINT coordinate and path-CV, yield free activation energies which are in good agreement with energies obtained using bond distance as CV. We believe that the current study will pave the way for further either short-term or long-term studies, leading to the novel discovery of and expanding the understanding of both iEDDA and azaphilic addition reactions.
References
Oliveira BL, Guo Z, Bernardes GJL (2017) Inverse electron demand Diels–Alder reactions in chemical biology. Chem Soc Rev 46(16):4895–4950. https://doi.org/10.1039/c7cs00184c
Zhang J, Shukla V, Boger DL (2019) Inverse electron demand Diels–Alder reactions of heterocyclic azadienes, 1-aza-1, 3-butadienes, cyclopropenone ketals, and related systems. a retrospective. J Org Chem 84(15):9397–9445. https://doi.org/10.1021/acs.joc.9b00834
Poty S, Membreno R, Glaser JM, Ragupathi A, Scholz WW, Zeglis BM, Lewis JS (2018) The inverse electron-demand Diels–Alder reaction as a new methodology for the synthesis of225ac-labelled radioimmunoconjugates. Chem Commun 54(21):2599–2602. https://doi.org/10.1039/c7cc09129j
Schnell SD, Schilling M, Sklyaruk J, Linden A, Luber S, Gademann K (2021) Nucleophilic attack on nitrogen in tetrazines by silyl-enol ethers. Org Lett 23(7):2426–2430. https://doi.org/10.1021/acs.orglett.0c04113
Ratnikov MO, Lipilin DL, Churakov AM, Strelenko YA, Tartakovsky VA (2002) Release of nitrosating species in the course of reduction of benzo-1, 2, 3, 4-tetrazine 1, 3-dioxides. Org Lett 4(19):3227–3229. https://doi.org/10.1021/ol026437o
Uhde M, Ziegler T (2010) Reaction of n-nitro-benzotriazole with nucleophiles. Synth Commun 40(20):3046–3057. https://doi.org/10.1080/00397910903353754
Jain S, Neumann K, Zhang Y, Valero E, Geng J, Bradley M (2016) Correction to tetrazine-mediated postpolymerization modification. Macromolecules 49(19):7608–7608. https://doi.org/10.1021/acs.macromol.6b02018
Knall A, Slugovc C (2013) Inverse electron demand Diels–Alder (iEDDA)-initiated conjugation: a (high) potential click chemistry scheme. Chem Soc Rev 42(12):5131. https://doi.org/10.1039/c3cs60049a
Pagel M (2018) Inverse electron demand Diels–Alder (IEDDA) reactions in peptide chemistry. J Pept Sci 25(1):e3141. https://doi.org/10.1002/psc.3141
Brinck T, Murray JS, Politzer P (1993) A computational analysis of the bonding in boron trifluoride and boron trichloride and their complexes with ammonia. Inorg Chem 32(12):2622–2625. https://doi.org/10.1021/ic00064a008
Jonas V, Frenking G, Reetz MT (1994) Comparative theoretical study of Lewis acid-base complexes of BH3, BF3, BCl3, AlCl3, and SO2. J Am Chem Soc 116(19):8741–8753. https://doi.org/10.1021/ja00098a037
Robinson EA, Johnson SA, Tang T, Gillespie RJ (1997) Reinterpretation of the lengths of bonds to fluorine in terms of an almost ionic model. Inorg Chem 36(14):3022–3030. https://doi.org/10.1021/ic961315b
Albert Cotton F, Albert Cotton F (eds) (1999) Advanced inorganic chemistry, 6th edition,. Wiley, New York
Fathimath Salfeena CT, Basavaraja Basavaraja KT, Ashitha VP, Kumar SV, Suresh CH, Sasidhar BS (2018) Synthesis of symmetrical and unsymmetrical triarylpyrylium ions via an inverse electron demand Diels–Alder reaction. Chem Commun 54(88):12463–12466. https://doi.org/10.1039/c8cc06444j
Cheng YS, Ho E, Mariano PS, Ammon HL (1985) Mechanistic aspects of the boron trifluoride catalyzed, intermolecular Diels–Alder cycloaddition of an unactivated 2-aza 1, 3-diene with electron-donating-substituted dienophiles. J Org Chem 50(26):5678–5686. https://doi.org/10.1021/jo00350a049
Wu H, Devaraj NK (2015) Inverse electron-demand Diels–Alder bioorthogonal reactions. Top Curr Chem 374(1):109–130. https://doi.org/10.1007/s41061-015-0005-z
Eising S, Engwerda AHJ, Riedijk X, Bickelhaupt FM, Bonger KM (2018) Highly stable and selective tetrazines for the coordination-assisted bioorthogonal ligation with vinylboronic acids. Bioconjug Chem 29(9):3054–3059. https://doi.org/10.1021/acs.bioconjchem.8b00439
Black K, Liu P, Xu L, Doubleday C, Houk KN (2012) Dynamics, transition states, and timing of bond formation in Diels–Alder reactions. Proc Natl Acad Sci 109(32):12860–12865. https://doi.org/10.1073/pnas.1209316109
Törk L, Jiménez-Osés G, Doubleday C, Liu F, Houk KN (2015) Molecular dynamics of the Diels–Alder reactions of tetrazines with alkenes and n2 extrusions from adducts. J Am Chem Soc 137(14):4749–4758. https://doi.org/10.1021/jacs.5b00014
Salavati-fard T, Caratzoulas S, Doren DJ (2015) DFT study of solvent effects in acid-catalyzed Diels–Alder cycloadditions of 2, 5-dimethylfuran and maleic anhydride. J Phys Chem A 119(38):9834–9843. https://doi.org/10.1021/acs.jpca.5b05060
Haghdadi M, Nab N, Bosra HG (2016) DFT study on the regio- and stereo-selectivity of the Diels–Alder reaction between a cycloprop-2-ene carboxylate and some cyclic 1, 3-dienes. Prog React Kinet Mech 41(2):193–204. https://doi.org/10.3184/146867816x14636767119354
Pestana LR, Hao H, Head-Gordon T (2019) Diels–Alder reactions in water are determined by microsolvation. Nano Lett 20(1):606–611. https://doi.org/10.1021/acs.nanolett.9b04369
Shrivastav G, Khan TS, Agarwal M, Ali Haider M (2018) A car-parrinello molecular dynamics simulation study of the retro Diels–Alder reaction for partially saturated 2-pyrones in water. J Phys Chem C 122(22):11599–11607. https://doi.org/10.1021/acs.jpcc.8b00250
Li P, Liu F, Jia X, Shao Y, Hu W, Zheng J, Mei Y (2018) Efficient computation of free energy surfaces of Diels–Alder reactions in explicit solvent at ab initio QM/MM level. Molecules 23(10):2487. https://doi.org/10.3390/molecules23102487
Torrie GM, Valleau JP (1977) Nonphysical sampling distributions in monte Carlo free-energy estimation: Umbrella sampling. J Comput Phys 23(2):187–199. https://doi.org/10.1016/0021-9991(77)90121-8
Laio A, Parrinello M (2002) Escaping free-energy minima. Proc Natl Acad Sci 99(20):12562–12566. https://doi.org/10.1073/pnas.202427399
Ensing B, De Vivo M, Liu Z, Moore P, Klein ML (2006) Metadynamics as a tool for exploring free energy landscapes of chemical reactions. Acc Chem Res 39(2):73–81. https://doi.org/10.1021/ar040198i
Piccini GM, Mendels D, Parrinello M (2018) Metadynamics with discriminants: a tool for understanding chemistry. J Chem Theory Comput 14(10):5040–5044
Carter EA, Ciccotti G, Hynes JT, Kapral R (1989) Constrained reaction coordinate dynamics for the simulation of rare events. Chem Phys Lett 156(5):472–477. https://doi.org/10.1016/s0009-2614(89)87314-2
Marx D, Hutter J (2009) Ab initio molecular dynamics. Cambridge University Press, Cambridge
Bucher D, Pierce LCT, McCammon JA, Markwick PRL (2011) On the use of accelerated molecular dynamics to enhance configurational sampling in ab initio simulations. J Chem Theory Comput 7(4):890–897. https://doi.org/10.1021/ct100605v
Paquet E, Viktor HL (2018) Computational methods for ab initio molecular dynamics. Adv Chem 2018:1–14. https://doi.org/10.1155/2018/9839641
Schilling M, Luber S (2019) Determination of pKa values via ab initio molecular dynamics and its application to transition metal-based water oxidation catalysts. Inorganics 7(6):73. https://doi.org/10.3390/inorganics7060073
Schilling M, Ketkaew R, Luber S (2021) How ab initio molecular dynamics can change the understanding on transition metal catalysed water oxidation. CHIMIA Int J Chem 75(3):195–201. https://doi.org/10.2533/chimia.2021.195
Grimme S, Bannwarth C, Shushkov P (2017a) A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (z= 1–86). J Chem Theory Comput 13(5):1989–2009
Ásgeirsson V, Bauer CA, Grimme S (2017) Quantum chemical calculation of electron ionization mass spectra for general organic and inorganic molecules. Chem Sci 8(7):4879–4895. https://doi.org/10.1039/c7sc00601b
Spicher S, Grimme S (2021) Single-point hessian calculations for improved vibrational frequencies and rigid-rotor-harmonic-oscillator thermodynamics. J Chem Theory Comput. https://doi.org/10.1021/acs.jctc.0c01306
Spicher S, Grimme S (2020) Efficient computation of free energy contributions for association reactions of large molecules. J Phys Chem Lett 11(16):6606–6611. https://doi.org/10.1021/acs.jpclett.0c01930
Bursch M, Hansen A, Pracht P, Kohn JT, Grimme S (2021) Theoretical study on conformational energies of transition metal complexes. Phys Chem Chem Phys 23(1):287–299. https://doi.org/10.1039/d0cp04696e
Spicher S, Bursch M, Grimme S (2020) Efficient calculation of small molecule binding in metal-organic frameworks and porous organic cages. J Phys Chem C 124(50):27529–27541. https://doi.org/10.1021/acs.jpcc.0c08617
Grimme S, Bannwarth C, Dohm S, Hansen A, Pisarek J, Pracht P, Seibert J, Neese F (2017b) Fully automated quantum-chemistry-based computation of spin-spin-coupled nuclear magnetic resonance spectra. Angew Chem Int Ed 56(46):14763–14769. https://doi.org/10.1002/anie.201708266
Seibert J, Bannwarth C, Grimme S (2017) Biomolecular structure information from high-speed quantum mechanical electronic spectra calculation. J Am Chem Soc 139(34):11682–11685. https://doi.org/10.1021/jacs.7b05833
de Wergifosse M, Grimme S (2018) Nonlinear-response properties in a simplified time-dependent density functional theory (sTD-DFT) framework: evaluation of the first hyperpolarizability. J Chem Phys 149(2):024108. https://doi.org/10.1063/1.5037665
de Wergifosse M, Grimme S (2019) Nonlinear-response properties in a simplified time-dependent density functional theory (sTD-DFT) framework: evaluation of excited-state absorption spectra. J Chem Phys 150(9):094112. https://doi.org/10.1063/1.5080199
Pietrucci F, Andreoni W (2014) Fate of a graphene flake: a new route toward fullerenes disclosed with ab initio simulations. J Chem Theory Comput 10(3):913–917. https://doi.org/10.1021/ct401103p
Zheng S, Pfaendtner J (2014) Car-parrinello molecular dynamics metadynamics study of high-temperature methanol oxidation reactions using generic collective variables. J Phys Chem C 118(20):10764–10770. https://doi.org/10.1021/jp500398k
CP2K Developers Group. Cp2k program package. https://www.cp2k.org/
Goedecker S, Teter M, Hutter J (1996) Separable dual-space gaussian pseudopotentials. Phys Rev B 54(3):1703–1710. https://doi.org/10.1103/physrevb.54.1703
Hartwigsen C, Goedecker S, Hutter J (1998) Relativistic separable dual-space gaussian pseudopotentials from h to rn. Phys Rev B 58(7):3641–3662. https://doi.org/10.1103/physrevb.58.3641
Krack M (2005) Pseudopotentials for h to kr optimized for gradient-corrected exchange-correlation functionals. Theoret Chem Acc 114(1–3):145–152. https://doi.org/10.1007/s00214-005-0655-y
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77(18):3865–3868. https://doi.org/10.1103/physrevlett.77.3865
Stefan G, Jens A, Stephan E, Helge K (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-d) for the 94 elements h-pu. J Chem Phys 132(15):3414. https://doi.org/10.1063/1.3382344
Tribello GA, Bonomi M, Branduardi D, Camilloni C, Bussi G (2014) PLUMED 2: new feathers for an old bird. Comput Phys Commun 185(2):604–613. https://doi.org/10.1016/j.cpc.2013.09.018
VandeVondele J, Krack M, Mohamed F, Parrinello M, Chassaing T, Hutter J (2005) Quickstep: fast and accurate density functional calculations using a mixed gaussian and plane waves approach. Comput Phys Commun 167(2):103–128. https://doi.org/10.1016/j.cpc.2004.12.014
...Balasubramani SG, Chen GP, Coriani S, Diedenhofen M, Frank MS, Franzke YJ, Furche F, Grotjahn R, Harding ME, Hättig C, Hellweg A, Helmich-Paris B, Holzer C, Huniar U, Kaupp M, Khah AM, Khani SK, Müller T, Mack F, Nguyen BD, Parker SM, Perlt E, Rappoport D, Reiter K, Roy S, Rückert M, Schmitz G, Sierka M, Tapavicza E, Tew DP, van Wüllen C, Voora VK, Weigend F, Wodyński A, Yu JM (2020) TURBOMOLE: modular program suite for ab initio quantum-chemical and condensed-matter simulations. J Chem Phys 152(18):184107. https://doi.org/10.1063/5.0004635
Perdew JP (1986) Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B 33(12):8822–8824. https://doi.org/10.1103/physrevb.33.8822
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38(6):3098–3100. https://doi.org/10.1103/physreva.38.3098
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for h to rn: design and assessment of accuracy. Phys Chem Chem Phys 7(18):3297. https://doi.org/10.1039/b508541a
Wang Y, Verma P, Jin X, Truhlar DG, He X (2018) Revised m06 density functional for main-group and transition-metal chemistry. Proc Natl Acad Sci 115(41):10257–10262. https://doi.org/10.1073/pnas.1810421115
Schäfer A, Klamt A, Sattel D, Lohrenz JCW, Eckert F (2000) COSMO implementation in TURBOMOLE: extension of an efficient quantum chemical code towards liquid systems. Phys Chem Chem Phys 2(10):2187–2193. https://doi.org/10.1039/b000184h
Alessandro B, Giovanni B, Michele P (2008) Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys Rev Lett 100(2):020603. https://doi.org/10.1103/physrevlett.100.020603
Ciccotti G, Ferrario M (2004) Blue moon approach to rare events. Mol Simul 30(11–12):787–793. https://doi.org/10.1080/0892702042000270214
Schilling M, Cunha RA, Luber S (2020) Zooming in on the o-o bond formation–an ab initio molecular dynamics study applying enhanced sampling techniques. J Chem Theory Comput 16(4):2436–2449. https://doi.org/10.1021/acs.jctc.9b01207
Dang LX (1999) Intermolecular interactions of liquid dichloromethane and equilibrium properties of liquid-vapor and liquid-liquid interfaces: a molecular dynamics study. J Chem Phys 110(20):10113–10122. https://doi.org/10.1063/1.478884
Yaws CL (1992) Thermodynamic and physical property data. Gulf Pub. Co., Houston
Rothschild WG (2007) Solvation by benzene: molecular dynamics simulation of orientational motion, translational diffusion, and site-site radial distributions of the solutes di- and trichloromethane (chloroform). Mol Phys 105(8):1003–1011. https://doi.org/10.1080/00268970701261431
Pfaendtner J, Bonomi M (2015) Efficient sampling of high-dimensional free-energy landscapes with parallel bias metadynamics. J Chem Theory Comput 11(11):5062–5067. https://doi.org/10.1021/acs.jctc.5b00846
Valsson O, Tiwary P, Parrinello M (2016) Enhancing important fluctuations: rare events and metadynamics from a conceptual viewpoint. Annu Rev Phys Chem 67(1):159–184. https://doi.org/10.1146/annurev-physchem-040215-112229
Pietrucci F, Andreoni W (2011) Graph theory meets Ab Initio molecular dynamics: atomic structures and transformations at the nanoscale. Phys Rev Lett 107(8):085504. https://doi.org/10.1103/physrevlett.107.085504
Fu CD, Pfaendtner J (2018) Lifting the curse of dimensionality on enhanced sampling of reaction networks with parallel bias metadynamics. J Chem Theory Comput 14(5):2516–2525. https://doi.org/10.1021/acs.jctc.7b01289
Branduardi D, Gervasio FL, Parrinello M (2007) From a to b in free energy space. J Chem Phys 126(5):054103. https://doi.org/10.1063/1.2432340
Neva B, Gervasio Francesco L (2011) Using metadynamics and path collective variables to study ligand binding and induced conformational transitions. In: Ambrosius WT (ed) Methods in molecular biology. Springer, New York, pp 501–513
de Alba Ortíz AP, Tiwari A, Puthenkalathil RC, Ensing B (2018) Advances in enhanced sampling along adaptive paths of collective variables. J Chem Phys 149(7):072320. https://doi.org/10.1063/1.5027392
Alba-Ortiz AP, Vreede J, Ensing B (2019) The adaptive path collective variable: a versatile biasing approach to compute the average transition path and free energy of molecular transitions. In: Ambrosius WT (ed) Methods in molecular biology. Springer, New York, pp 255–290
Leines GD, Ensing B (2012) Path finding on high-dimensional free energy landscapes. Phys Rev Lett 109(2):020601. https://doi.org/10.1103/physrevlett.109.020601
Marcos-Alcalde I, Setoain J, Mendieta-Moreno JI, Mendieta J, Gómez-Puertas P (2015) MEPSA: minimum energy pathway analysis for energy landscapes. Bioinformatics 3853–3855. https://doi.org/10.1093/bioinformatics/btv453
Hodel FH, Deglmann P, Luber S (2017) Exploring solvation effects in ligand-exchange reactions via static and dynamic methods. J Chem Theory Comput 13(7):3348–3358. https://doi.org/10.1021/acs.jctc.7b00214
Schilling M, Hodel FH, Luber S (2017) Discovery of open cubane core structures for biomimetic LnCo3 (OR)4 water oxidation catalysts. Chemsuschem 10(22):4561–4569. https://doi.org/10.1002/cssc.201701527
Widstrom AL, Lear BJ (2019) Structural and solvent control over activation parameters for a pair of retro Diels–Alder reactions. Sci Rep 9(1):1–8. https://doi.org/10.1038/s41598-019-54156-4
Acknowledgements
This work is supported by the University of Zurich and the Swiss National Science Foundation (Grant No. PP00P2_170667). The authors thank the Swiss National Supercomputing Centre for computational power support (Project I.D. s875, s1001, and s1036). We thank Mauro Schilling for help in preparing and carrying out the calculations, analysis, and discussions.
Funding
Open Access funding provided by Universität Zürich.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ketkaew, R., Creazzo, F. & Luber, S. Closer Look at Inverse Electron Demand Diels–Alder and Nucleophilic Addition Reactions on s-Tetrazines Using Enhanced Sampling Methods. Top Catal 65, 1–17 (2022). https://doi.org/10.1007/s11244-021-01516-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-021-01516-y