Adding to the Family of Copper Complexes Featuring Borohydride Ligands Based on 2-Mercaptopyridyl Units
by
 , , and
, , and
Joseph Goldsworthy
1, 
Simon D. Thomas
1,
Graham J. Tizzard
2,
Simon J. Coles
2 and
Gareth R. Owen
1,* 1
School of Applied Sciences, University of South Wales, Pontypridd CF37 4AT, UK
2
UK National Crystallography Service, University of Southampton, Highfield, Southampton SO17 1BJ, UK
*
Author to whom correspondence should be addressed.
Inorganics 2019, 7(8), 93; https://doi.org/10.3390/inorganics7080093
Submission received: 13 June 2019
/
Revised: 18 July 2019
/
Accepted: 19 July 2019
/
Published: 24 July 2019
(This article belongs to the Special Issue Metal Complexes Containing Boron Based Ligands)
Abstract
:Borohydride ligands featuring multiple pendant donor functionalities have been prevalent in the chemical literature for many decades now. More recent times has seen their development into new families of so-called soft scorpionates, for example, those featuring sulfur based donors. Despite all of these developments, those ligands containing just one pendant group are rare. This article explores one ligand family based on the 2-mercaptopyridine heterocycle. The coordination chemistry of the monosubstituted ligand, [H3B(mp)]− (mp = 2-mercaptopyridyl), has been explored. Reaction of Na[BH3(mp)] with one equivalent of Cu(I)Cl in the presence of either triphenylphosphine or tricyclohexylphosphine co-ligands leads to the formation of [Cu{H3B(mp)}(PR3)] (R = Ph, 1; Cy, 2), respectively. Structural characterization confirms a κ3-S,H,H coordination mode for the borohydride-based ligand within 1 and 2, involving a dihydroborate bridging interaction (BH2Cu) with the copper centers.
1. Introduction
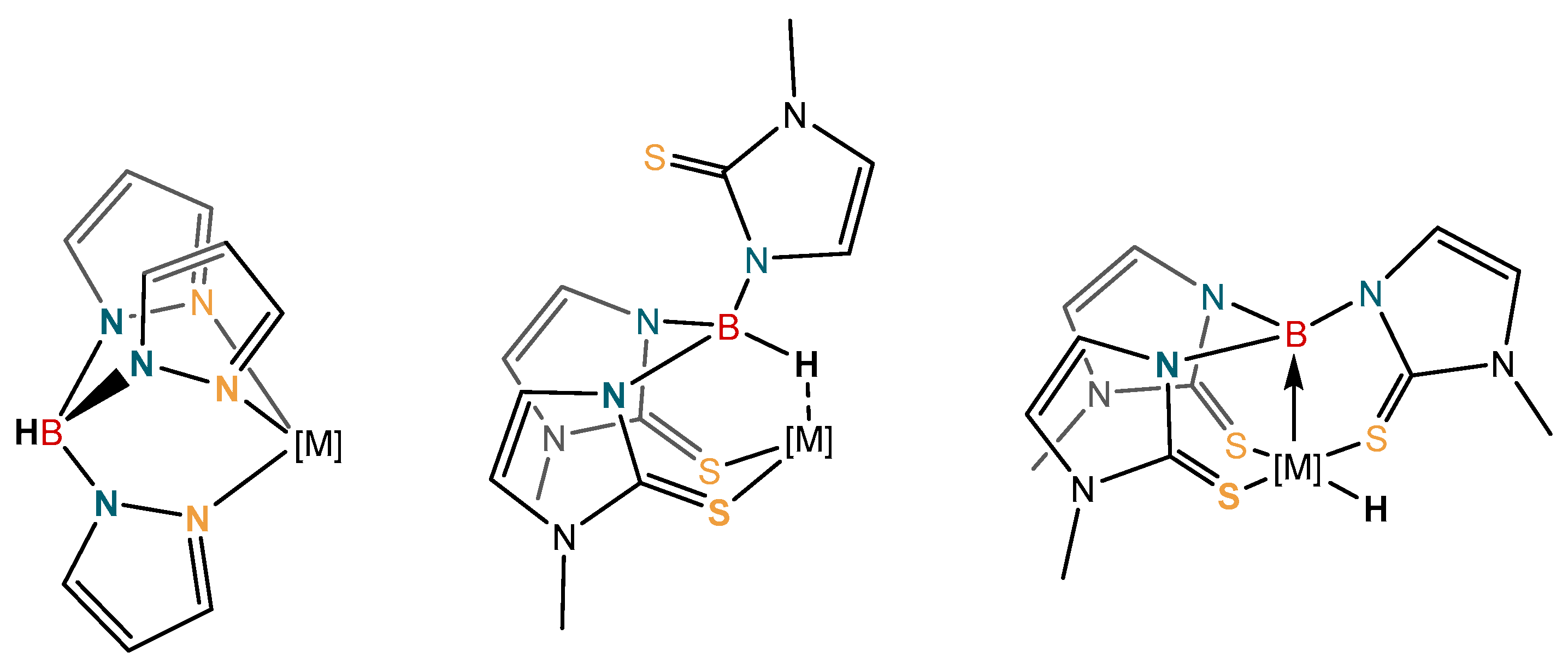
The coordination chemistry of borohydride and substituted borohydride units with transition metals has been a major focus of research over many decades now [1,2,3,4,5,6,7,8,9]. One particular focus of research has been on substituted borohydride units attached to other donor functional groups. This gave rise to a research area known as “scorpionate chemistry”, where the borohydride moiety is typically substituted by two or more pyrazolyl rings, thus forming multidentate ligand systems. This area of research has provided an expansive and fascinating array of compounds with wide ranging applications. These have been explored in homogeneous catalysis and bioinorganic chemistry, for example [10,11,12,13,14,15,16]. In many examples, the borohydride unit is positioned away from the metal center, playing a spectator role within the complex. The polyprazolylborates, for example, are known as “octahedral enforcers”, furnishing highly rigid stable complexes.
More recent developments, have led to new generations of ligand systems, where the borohydride unit is positioned in direct contact with the metal center. In some cases these can undergo direct transformations at the boron center (Figure 1) [17,18,19,20,21,22]. This occurs when an additional atom is incorporated between the boron and donor atom. The publication of this new generation of more “flexible scorpionates” opened up a new area of research with respect to the formation of Z-type ligands [17,18,19,20,21,22]. This revolutionized the field and altered the perspective on the coordination chemistry of such ligands. The first of the more flexible scorpionate ligands was [Tm]− [hydrotris(methylimidazolyl)borate] (Figure 1; middle) [23]. This new ligand had two major differences when compared to Trofimenko’s original scorpionates. The ligand was based on soft sulfur donor atoms [16], and perhaps more significantly, greater flexibility had been incorporated into the ligand by addition of the extra atom between the boron and the donor atom. It was this greater flexibility within the ligand structure that opened up the potential for activation at the boron bridgehead and formation of metal-borane (metallaboratrane) complexes [17,18,19,20,24,25,26,27], giving rise to reactivity not observed in the analogous polypyrazolylborate ligands [10,11,12,13,14,15,16].
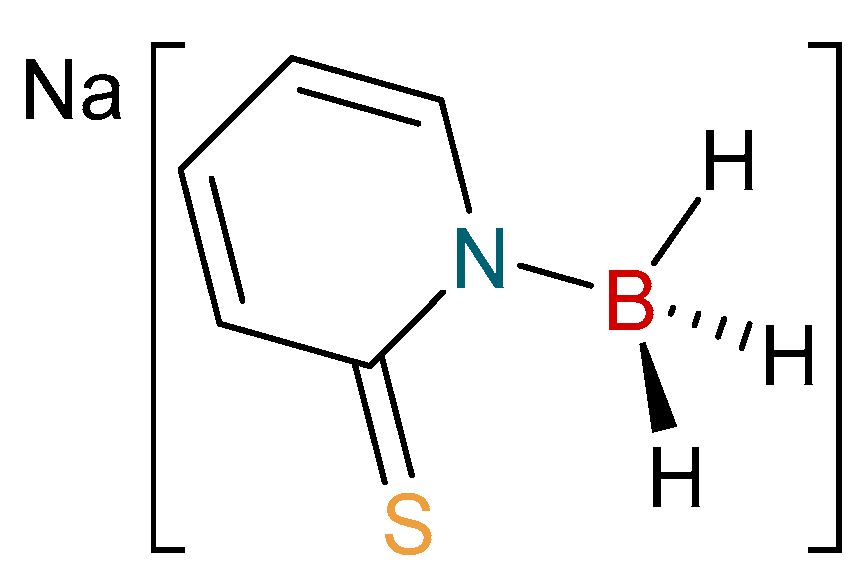
Over the following twenty years since the first report of hydride migration from the boron center of a scorpionate ligand, a number of research groups have focused on new, more flexible borohydride ligands containing a range of supporting units based on nitrogen [28,29,30,31] and other sulfur heterocycles [32,33,34,35,36,37,38,39,40,41,42,43,44,45,46]. As part of our research, we have focused on providing new derivative ligand systems. In 2009, we introduced a new family of flexible scorpionate ligands derived from the 2-mercaptopyridine heterocycle [36]. This original report provided a borohydride-based ligand substituted by two and three of these heterocycles. Last year, we extended this family to include the monosubstitued ligand, [H3B(mp)]− (where mp = 2-mercaptopyridyl; Figure 2) [37]. Herein, we report the synthesis and characterization of the first copper complexes containing this new ligand. The complexes have been structurally characterized and compared to related complexes.
2. Results and Discussion
2.1. Synthesis and Characterization of Copper Complexes
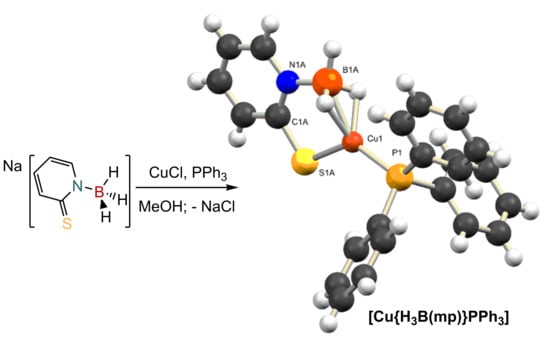
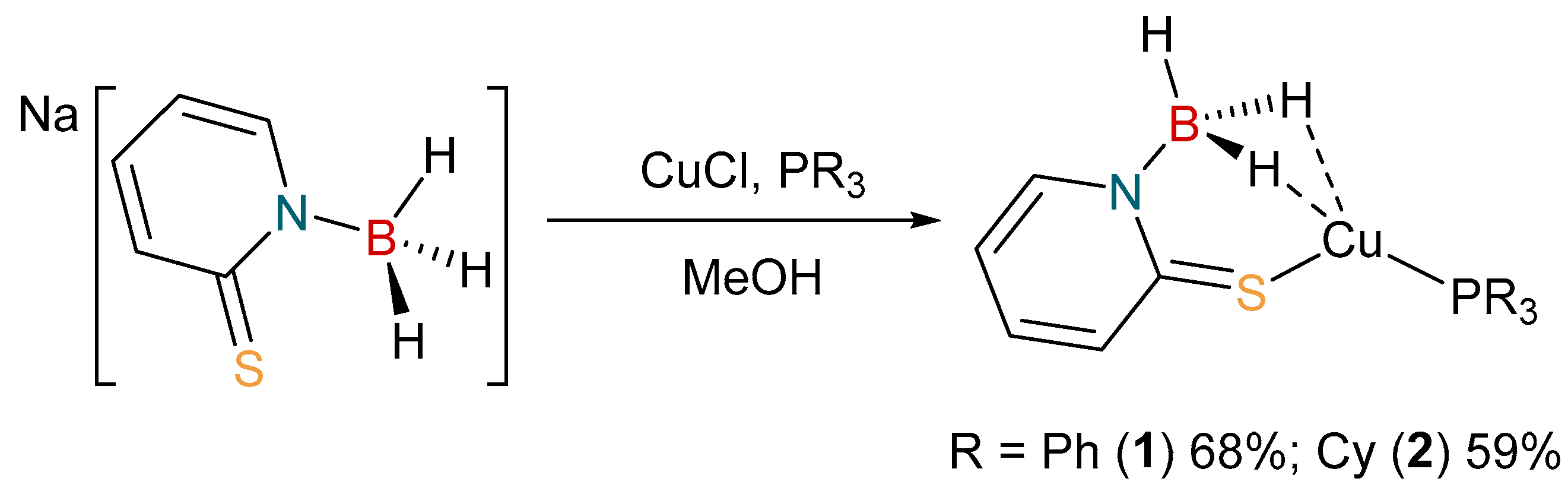
The coordination chemistry of [H3B(mp)]− is limited to one example to date. The complex [Rh{κ3-B,H,H-H3B(mp)}(NBD)] (where NBD = 2,5-norbornadiene), was reported by us in 2018 [37]. Accordingly, we set out to prepare some further examples of complexes containing this ligand. We have previously synthesized a series of copper(I) complexes containing the bis- and tris-substituted derivatives [36]. A similar synthetic protocol was, therefore, undertaken to prepare the complexes, [Cu{H3B(mp)}(PR3)] (R = Ph, 1; Cy, 2), as shown in Scheme 1. These complexes were readily prepared by reaction of one equivalent of Na[H3B(mp)] with one equivalent of CuCl in the presence of a stoichiometric amount of the corresponding phosphine co-ligand. The reactions were performed in methanol solvent, from which the products precipitated out as yellow solids.
The air stable products were obtained in good yields and were fully characterized by NMR and IR spectroscopy, mass spectrometry, and elemental analysis. Selected characterization data for complexes 1 and 2 are presented in Table 1, along with data for the corresponding copper complexes containing the bis- and tris-substituted ligands, [H2B(mp)2]− and [HB(mp)3]−, for comparison. The 11B NMR spectra of complexes 1 and 2, in CDCl3, revealed single broad resonances at −13.9 ppm and −13.4 ppm, respectively (see Figures S3 and S10 in the Supplementary Materials). Both signals presented as poorly unresolved quartets with 1JBH coupling constants of 75 Hz for 1 and 82 Hz for 2. Both were found to be singlet resonances in the corresponding 11B{1H} NMR spectra (with half height widths 113 Hz and 90 Hz, respectively), confirming that three hydrogen substituents remain at the boron center. The change in chemical shift from the starting material to the complexes was insignificant (c.f. −14.1 ppm in CD3CN), particularly when taking into account the different solvent. There does seem to be a small reduction in the 1JBH coupling constant upon coordination of [H3B(mp)]− to the copper center. In Na[H3B(mp)], this value is 93 Hz. From these data, it appears that the BH3 unit of the ligand is not strongly interacting with the copper metal center. Similar observations have been reported for neutral borane adducts, of the type H3BNR3, with copper complexes [47,48]. This is in contrast to those observations for [Rh{κ3-B,H,H-H3B(mp)}(NBD)], in which the boron chemical shift in complexes was found to be −7.8 ppm. As highlighted in Table 1, the change in boron chemical shift upon complexation was a little more pronounced for the copper complexes bearing the [H2B(mp)2]− and [HB(mp)3]− ligands.
Further information on these complexes was obtained from their 31P{1H} NMR spectra. The 31P{1H} NMR spectra of 1 and 2 revealed single broad resonances at 4.8 ppm and 27.2 ppm, respectively (Figures S6 and S13). These both represent downfield chemical shifts with respect to the free phosphines, confirming their coordination to the metal centers. These changes in chemical shift with respect to the free phosphines are more significant than the corresponding bis- and tris-complexes, suggesting that the phosphines are more strongly bound in the lower coordination complexes, as might be expected. As indicated above, the 11B NMR data did not unambiguously confirm coordination of H3B(mp) unit. The 1H NMR data, on the other hand, were a little more convincing, exhibiting a new set of signals for the mercaptopyridyl protons with clear shifts from the starting material. The 1H NMR spectrum for 1 (Figure S1) showed an integration ratio of 3H:16H:1H:1H:1H corresponding to the BH3 group, 15 aromatic protons on the triphenylphosphine ligand, plus one overlapping proton environment on the mercaptopyridine unit. The three remaining signals corresponded to the other proton environments on the mercaptopyridyl unit. A similar situation was found for complex 2, confirming the presence of the BH3 unit, the mercaptopyridyl heterocycle, and the PCy3 ligand within the complex (Figure S8). For both complexes, the BH3 protons were located at significantly broad signals at 2.64 ppm for 1 and 2.42 ppm for 2, in their 1H{11B} NMR spectra. These were shifted downfield with respect to [H3B(mp)]−, which were observed at 2.11 ppm. Again, the corresponding shifts for [Rh{κ3-B,H,H-H3B(mp)}(NBD)] were −2.72 ppm (integrating for 2 H) and 2.89 ppm (integrating for 1 H) for the bridging and terminal hydrogen substituents on boron. This, of course, represents a static BH2 bridging interaction with the rhodium center, whereas a fluxional interaction must be present in complexes 1 and 2, since all three hydrogens at boron are in the same chemical environment. A series of 13C{1H} and two-dimensional correlation NMR experiments were carried out to fully assign all hydrogen and carbon chemical environments within the two complexes (see Experimental section). Further evidence of coordination of [H3B(mp)]− to the metal center was found in the infrared spectrum. Powder film samples gave characteristic bands at 2439 cm−1 for 1 and 2448 cm−1 for 2, corresponding to the terminal B‒H stretch. These compared to the 2307 cm−1 value found for Na[H3B(mp)] [37]. Two additional bands were also located in the IR spectra for 1 and 2 at 2078 cm−1 and 2085 cm−1, respectively. These correspond to the BH2Cu interactions, where two of the three B‒H bonds in the BH3 unit interact with the metal center [1,2,3,4]. The crystal structure previously reported for [Cu{H2B(mp)2}PPh3] contains a κ3-S,S,H coordination mode for the scorpionate ligand, involving the interaction of one of the B‒H bonds with the copper center [36]. This is presumably due to the preference for coordination of an additional sulfur donor to the metal center over the BH2Cu mode and the restriction against a κ4-S,S,H,H coordination mode. The compounds were also analyzed by mass spectrometry. The molecular ion peak was found for 2 by mass spectrometry. For complex 1, only the fragment [Cu(mpH)(PPh3)] was observed. Finally, confirmation for the formation of the targeted products was confirmed by satisfactory elemental analysis.
2.2. Structural Characterization of Copper Complexes
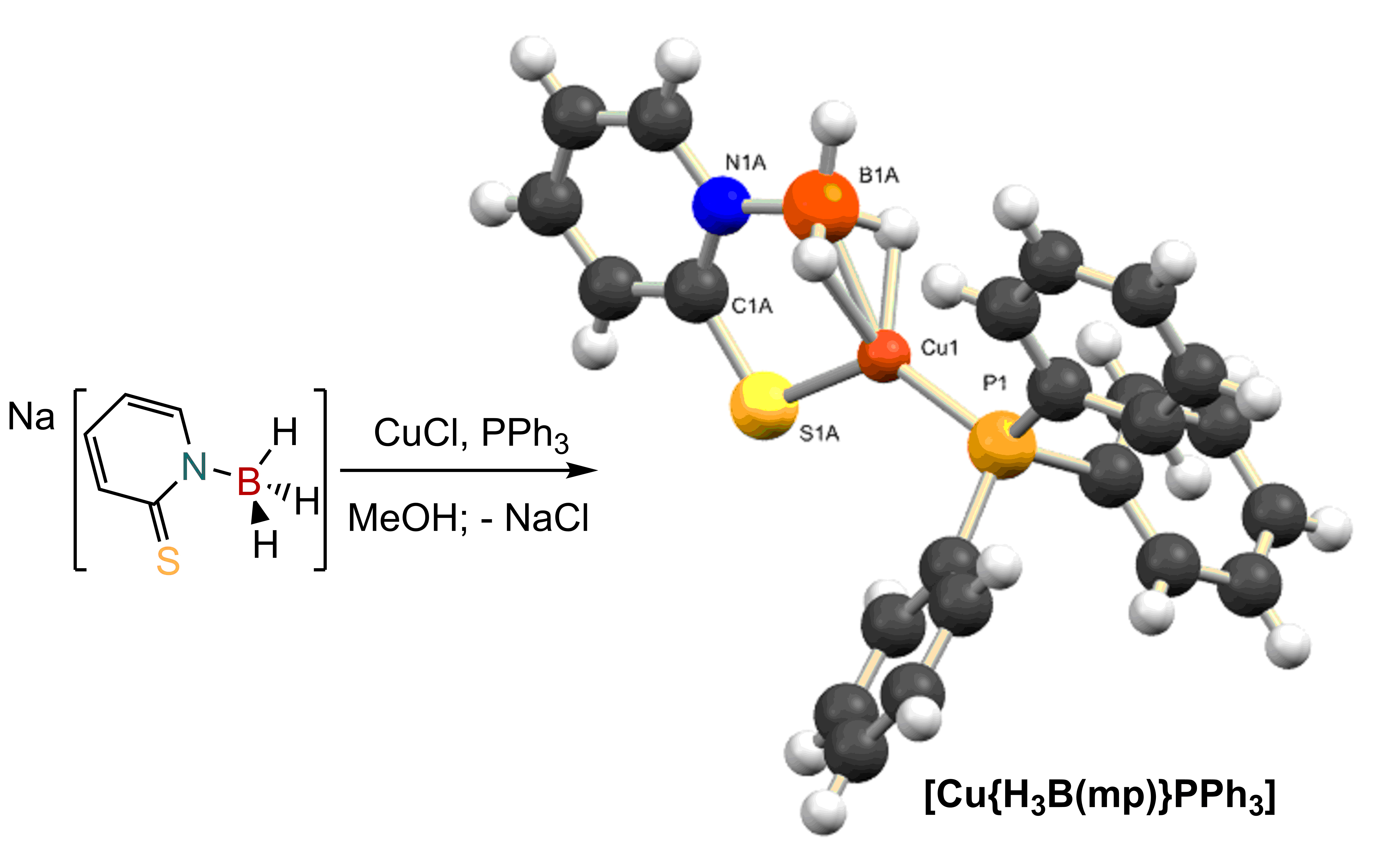
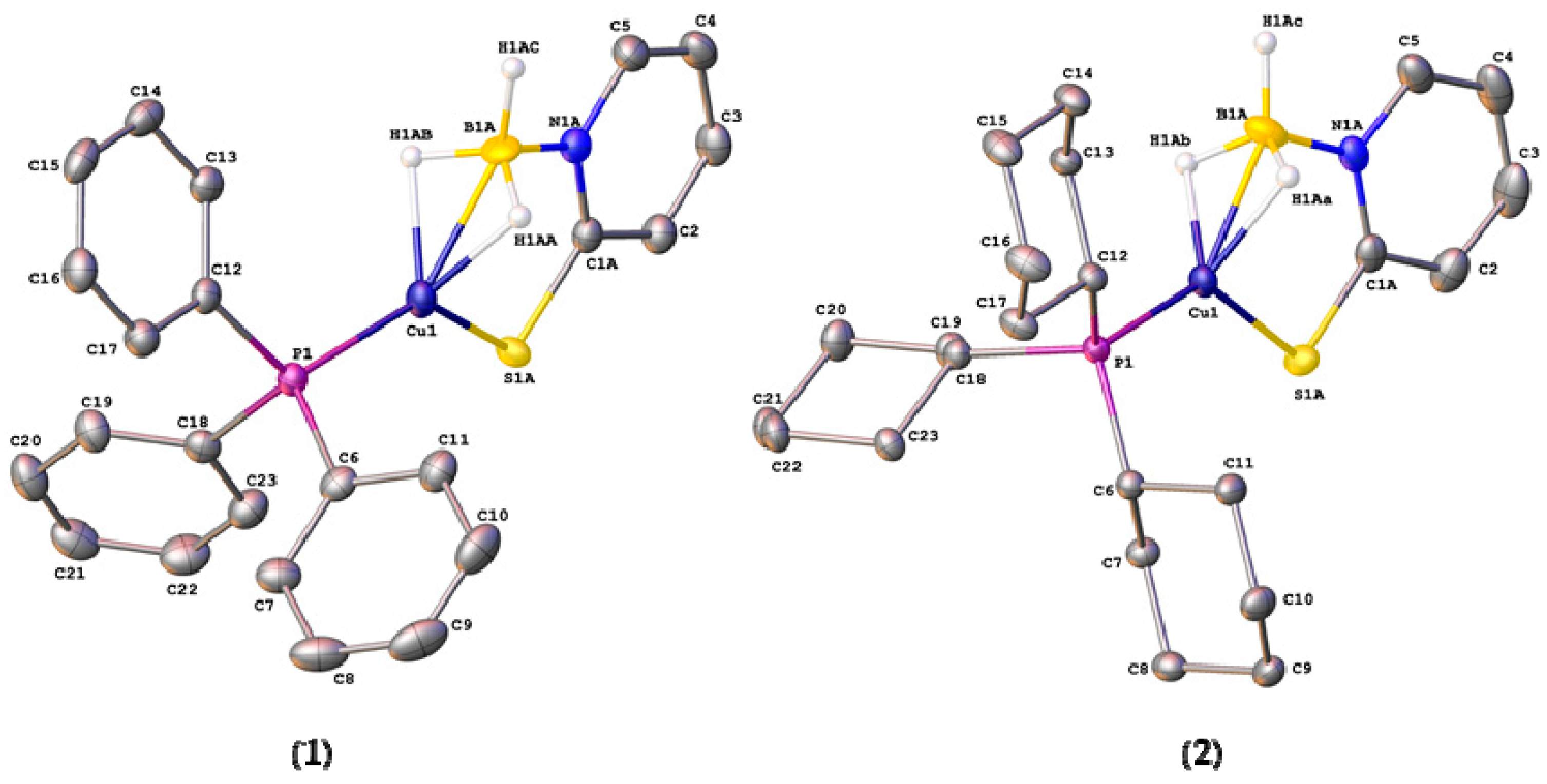
Single crystals of complexes 1 and 2, suitable for X-ray crystallography, were obtained from slow evaporation of the solvent from diethyl ether—methanol (1:1) mixtures. The molecular structures of these complexes are shown in Figure 3. Selected bond distances and angles for these complexes are shown in Table 2, along with those for [Cu{H2B(mp)2}PPh3] (3) for comparison. Crystallographic parameters are provided in the supporting information. The two new structures contained disorder in the position of the [H3B(mp)]− ligand in ratios 56:44 for complex 1 and 79:21 for complex 2. The lack of strong H-bond donor/acceptors in either complex results in simple close-packed crystal structures driven by dispersion forces. The structures of both 1 and 2 confirmed the coordination of one phosphine ligand and one [H3B(mp)]− ligand to the metal center. The solid state structures confirmed that the BH3 unit was bound to the copper center via a BH2Cu bridging mode. This is, therefore, consistent with the IR spectroscopic data. The BH2Cu mode can either be considered as two separate B‒H agostic type interactions (η2,η2) or as a three-centered dihydroborate interaction [1,2,3,4]. This coordination mode in the mono-substituted ligand allows for a different morphology about the copper center in comparison to that found in complex 3 [36]. If the hydrogen substituents are ignored and the boron center of the BH3 unit is considered as the site of coordination at the copper, then the geometries around the metal center are highly distorted trigonal planar structures. In both cases, if a plane is defined by the atoms P(1), B(1), and S(1), then the copper center sits in a position that is very close to this plane. The distance of the copper center from these planes is 0.062(7) Å for 1 and 0.019(6) Å for 2. The sums of the aforementioned angles are very close to the idealized 360°. The ligand forms a six-membered ring where it links to the copper via the sulfur donor and the hydrogen substituents at boron. Whilst the BH2Cu interaction does not appear to be strong in solution, it appears that the BH3 unit is held in close proximity to the metal center via the mercaptopyridine supporting unit. In the case of [Cu{κ3-S,S,H-H2B(mp)2}PPh3], a distorted geometry between tetrahedral and trigonal pyramidal is observed as demonstrated by the sum of the same angles, which is 350.4°. In this complex, two six-membered rings are formed as a result of the κ3-S,S,H coordination mode. In the absence of the BHCu interaction, this would have led to formation of one eight membered ring.
The Cu(1)–B(1) distances in complexes 1 are 2.113(17) Å (major component in disorder) and 2.229(14) Å (minor component). The corresponding distances in 2 are 2.153(16) Å and 2.10(3) Å, respectively. These distances are consistent with similar copper complexes featuring a neutral H3BN moiety bound to the metal center with a dihydroborate mode [46,47]. Again, the difference in the coordination mode from κ3-S,H,H in 1 and 2 to κ3-S,S,H in 3 is significant. In complex 3, the Cu‒B distance is 2.7479(15) Å, since this represents a Cu‒H‒B bridging interaction. The Cu(1)–S(1) distances for complex 1 are 2.205(2) Å (major) and 2.221(4) Å (minor). For complex 2, the corresponding Cu(1)–S(1) distances are 2.2523(12) Å and 2.296(12) Å. This indicates that the interaction of the thione unit with the metal center in 2 is weaker than in 1, as might be expected, since complex 2 contains the more electron-rich phosphine ligand.
The B‒N and C‒S distances within the complexes are of interest in order to explore the extent of different resonance forms within the [H3B(mp)]− ligand. The ligand can be described as a thiopyridone species forming a borohydride entity (Figure 4, left), or as a pyridine-2-thiolate forming a borane adduct (Figure 4, right). As can be observed in Table 2, the B‒N and C‒S distances vary significantly within the disordered components of the complexes. For example, in complex 1 the C(1)–S(1) distances are 1.7515(17) Å (for the major component of disorder) and 1.722(2) Å (for the minor). The former represents a significant difference in bond order between single and double bond character. It is interesting to note that the corresponding distances in the previously reported complex, [Cu{H2B(mp)2}PPh3], are shorter, suggesting a more double-bonded character in the bis-substituted ligand.
3. Materials and Methods
3.1. General Remarks
The syntheses of the complexes were carried out using standard Schlenk techniques. Solvents were sources as extra dry from “Acros Organics” (Morris Plains, NJ, USA) and were stored over either 4 Å or 3 Å molecular sieves. The NMR solvent, CDCl3, was stored in Young’s ampule over 4 Å molecular sieves, under a N2 atmosphere and was degassed through freeze–thaw cycles prior to use. Reagents were used as purchased from commercial sources. The ligand Na[H3B(mp)] [36] was synthesized according to standard literature procedures. NMR spectroscopy experiments were conducted on a Bruker 400 MHz AscendTM 400 spectrometer (Billerica, MA, USA). All spectra were referenced internally, to the residual protic solvent (1H) or the signals of the solvent (13C). Proton (1H) and carbon (13C) assignments were further supported by heteronuclear single-quantum correlation spectroscopy (HSQC), heteronuclear multiple-bond correlation spectroscopy (HMBC), and correlation spectroscopy (COSY) two-dimensional correlation NMR experiments. The symbol “τ” is used to represent an apparent triplet, where the resonance is expected to be a “dd”. In these cases, the apparent coupling constant has been provided. Infrared spectra were recorded on a PerkinElmer Spectrum Two Attenuated total reflectance infra-red (ATR FT-IR) spectrometer as powder films (Foster City, CA, USA). Elemental analysis was performed at London Metropolitan University by their elemental analysis service. Mass spectra were recorded by the EPSRC National Mass Spectrometry Facility (NMSF) at Swansea University. The numbering scheme used for NMR assignments is highlighted in Figure 5.
3.2. Synthesis of [Cu{H3B(mp)}(PPh3)]
To a Schlenk flask containing CuCl (24 mg, 0.24 mmol), PPh3 (117 mg, 0.45 mmol), and Na[H3B(mp)] (33 mg, 0.22 mmol) was added methanol (5 mL). The stirred solution gradually turned yellow and a precipitate formed. The reaction was left stirring for 36 h, after which the flask was cooled to −40 °C and left overnight to further precipitate the product out of solution. The filtrate was removed via cannula filtration and the resultant solid dried under vacuum to give [Cu{H3B(mp)} (PPh3)] as a pale yellow powder (68 mg, 0.14 mmol, 68%).
1H NMR (δ, CDCl3): 6.76 (1H, τ, JHH = 6.5 Hz, mpCH-(4)), 7.17–7.44 (16H, m, P(C6H5)3 + mpCH-(5) [49]), 7.80 (1H, d, 3JHH = 8.5 Hz, mpCH-(6)), 8.51 (1H, d, 3JHH = 5.8 Hz mpCH-(3)). 1H{11B} (δ, CDCl3): 2.64 (3H, s br, BH3). 13C{1H} (δ, CDCl3): 115.6 (mpCH-(4)), 128.6 (d, 2JCP = 9.6 Hz, Portho(C6H5)3), 130.0 (d, 4JCP = 1.5 Hz, Ppara(C6H5)3), 131.5 (mpCH-(6)), 132.9 (d, 1JCP = 32 Hz, Pipso(C6H5)3), 133.8 (d, 3JCP = 16 Hz, Pmeta(C6H5)3), 135.0 (mpCH-(5)), 146.5 (mpCH-(3)), 175.9 (mpC=S-(2)). 31P{1H} NMR (δ, CDCl3): 4.8 (s, h.h.w. = 392 Hz). 11B NMR (δ, CDCl3): −13.9 (q, 1JBH = 75 Hz, BH3). 11B{1H} NMR (δ, CDCl3): −13.9 (s, h.h.w. = 113 Hz). MS APCI (ASAP+) m/z = 436.03 [M – BH3 + H]+. IR (cm−1, powder film) 2439 w (B–H), 2078 w (BH2Cu), 1614 s, 1568 s. Elemental analysis (%): Calculated for CuSNPC23H22B: C 61.41 H 4.93 N 3.11 Found: C 61.56 H 4.80 N 3.15.
3.3. Synthesis of [Cu{H3B(mp)}(PCy3)]
To a Schlenk flask containing CuCl (22 mg, 0.22 mmol), PCy3 (123 mg, 0.44 mmol), and Na[H3B(mp)] (30 mg, 0.20 mmol) was added methanol (5 mL). The stirred solution gradually turned yellow and a precipitate formed. The reaction was left stirring for 36 h, after which the flask was cooled to −40 °C and left overnight to further precipitate the product out of solution. The filtrate was removed via cannula filtration and the resultant solid dried under vacuum to give [Cu{H3B(mp)} (PCy3)] as an off white powder (62 mg, 0.14 mmol, 59%).
1H NMR (δ, CDCl3): 1.19–1.35 (21H, m, PCy3), 1.64–1.87 (23H, m, PCy3), 2.42 (3H, d vb, 1JBH = 106 Hz, BH3), 6.71 (1H, τ, JHH = 6.6 Hz, mpCH-(3)), 7.29 (1H, τ, JHH = 7.6 Hz, mpCH-(4)), 7.75 (1H, d, J = 8.3 Hz, mpCH-(5)), 8.48 (1H, d, J = 6.3 Hz, mpCH-(6)). 1H{11B} NMR (δ, CDCl3): 2.42 (3H, s br, BH3). 13C{1H} (δ, CDCl3): 26.2 (PCy3-(4)), 27.4 (d, 3JCP = 11 Hz, PCy3-(3)), 30.6 (d, 2JCP = 4 Hz, PCy3-(2)), 31.8 (d, 1JCP = 18 Hz, PCy3-(1)), 115.3 (mpCH-(4)), 131.4 (mpCH-(6)), 134.8 (mpCH-(5)), 146.3 (mpCH-(3)), 176.1 (mpC=S-(2)). 31P{1H} NMR (δ, CDCl3): 27.2 (s br, h.h.w. = 111 Hz). 11B NMR (δ, CDCl3): −13.4 (q, 1JBH = 82 Hz, BH3). 11B{1H} NMR (δ, CDCl3) −13.4 (s, h.h.w. = 90 Hz). IR (cm−1, powder film) 2448 w (B–H), 2085 w (BH2Cu), 1606 s, 1540 s. MS APCI (ASAP+) m/z = 467.2 [M]+. Elemental analysis (%): Calculated for C23H40BCuSNP: C 59.03 H 8.62 N 2.99, Found: C 59.21 H 8.48 N 2.90.
3.4. Crystallography
Single-crystal X-ray diffraction studies of complexes 1 and 2 were undertaken at the U.K. National Crystallography Service (NCS) at the University of Southampton [50]. Single crystals of each of the complexes were obtained by allowing a 1:1 mixture of methanol and diethyl ether to slowly evaporate at room temperature. For each sample, single crystal was mounted on a MITIGEN holder in perfluoroether oil on a Rigaku FRE+ equipped with HF Varimax confocal mirrors and an AFC11 goniometer and HyPix 6000 detector. The data for the crystals was collected at T = 100(2) K. Data were collected and processed via standard protocols. Empirical absorption corrections were carried out using CrysAlisPro [51]. The structures were solved by Intrinsic Phasing using the ShelXT structure solution program [52] and refined on Fo2 by full-matrix least squares refinement with version 2018/3 of ShelXL [53], as implemented in Olex2 [54]. All hydrogen atom positions, with the exception of those at boron, were calculated geometrically and refined using the riding model. Crystal Data for 1. C23H22BCuNPS, Mr = 449.79, monoclinic, C2/c (No. 15), a = 11.90994(6) Å, b = 13.21619(7) Å, c = 26.83905(13) Å, β = 97.6274(4)°, α = γ = 90°, V = 4187.20(4) Å3, T = 100(2) K, Z = 8, Z’ = 1, μ(Cu Kα) = 3.175 mm−1, 38,239 reflections measured, 3963 unique (Rint = 0.0259), which were used in all calculations. The final wR2 was 0.0664 (all data) and R1 was 0.0244 (I > 2(I)). Crystal Data for 2. C23H40BNPSCu, Mr = 467.94, triclinic, P-1 (No. 2), a = 8.16720(10) Å, b = 9.38370(10) Å, c = 17.2612(2) Å, α = 96.9390(10)°, β = 95.6170(10)°, γ = 112.3730(10)°, V = 1199.33(3) Å3, T = 100(2) K, Z = 2, Z’ = 1, μ(Cu Kα) = 2.773 mm−1, 30,937 reflections measured, 4471 unique (Rint = 0.0278), which were used in all calculations. The final wR2 was 0.0628 (all data) and R1 was 0.0236 (I > 2(I)). A summary of the crystallographic data collection parameters and refinement details for the complexes are presented in the supplementary information. Anisotropic parameters, bond lengths, and (torsion) angles for these structures are available from the CIF files, which have been deposited with the Cambridge Crystallographic Data Centre and given the following deposition numbers, 1922838 (1) and 1922839 (2). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
4. Conclusions
The synthesis and characterization of the first examples of copper complexes containing the mono-substituted borohydride ligand, [H3B(mp)]−, have been reported. These add to the family of ligands in which the bis- and tri-substituted versions have previously been reported. Mono-substituted soft borohydride derivatives are a rare class of compound and these examples are an interesting addition to the family. The new complexes were also structurally characterized by X-ray crystallography, which confirmed the κ3-S,H,H coordination mode where the BH3 unit coordinated via a BH2Cu bridging mode. The spectroscopic data appears to suggest the coordination of this unit to the metal center is weak in the case of copper. This is in contrast to a much stronger interaction that was found in the previously reported complex, [Rh{κ3-H,H,S-H3B(mp)}(NBD)]. The additional knowledge on the coordination chemistry of mono-substituted ligand systems, particularly the nature of the BH2Cu bridging mode, is of value.
Supplementary Materials
The following are available online at https://www.mdpi.com/2304-6740/7/8/93/s1. Table S1—crystallographic parameters for 1 and 2; Figures S1–S13—NMR spectra for complexes 1 and 2; CIF file and checkCIF file—crystallographic data for 1 and 2.
Author Contributions
J.G. and S.D.T. performed the experiments. G.J.T. and S.J.C. carried out the crystallography work. G.R.O. wrote the manuscript and directed the project.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Marks, T.J.; Kolb, J.R. Covalent transition metal, lanthanide, and actinide tetrahydroborate complexes. Chem. Rev. 1977, 77, 263–293. [Google Scholar] [CrossRef]
- Bommer, J.C.; Morse, K.W. Slowing of the fluxional process in a diamagnetic copper(I) tetrahydroborate complex. Inorg. Chem. 1978, 17, 3708–3710. [Google Scholar] [CrossRef]
- Aqra, F.M.A.M. Bidentate bonding mode of tetrahydroborate and nitrite towards copper(II) in open-faced macrocyclic complexes. Trans. Met. Chem. 2004, 29, 921–924. [Google Scholar] [CrossRef]
- Golub, I.E.; Filippov, O.A.; Gutsul, E.I.; Belkova, N.V.; Epstein, L.M.; Rossin, A.; Peruzzini, M.; Shubina, E.S. Dimerization Mechanism of Bis(triphenylphosphine)copper(I) Tetrahydroborate: Proton Transfer via a Dihydrogen Bond. Inorg. Chem. 2012, 51, 6486–6497. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Lin, Z. Transition metal tetrahydroborato complexes: An orbital interaction analysis of their structure and bonding. Coord. Chem. Rev. 1996, 156, 139–162. [Google Scholar] [CrossRef]
- Lledos, A.; Duran, M.; Jean, Y.; Volatron, F. Ab initio Study of the coordination modes of tetrahydroborato ligands: The high-spin complex bis(phosphine)tris(tetrahydroborato)vanadium. Inorg. Chem. 1991, 30, 4440–4445. [Google Scholar] [CrossRef]
- Dias, H.V.R.; Lu, H.-L. Direct Synthesis of a Bis(pyrazolyl)boratocopper(I) Complex: Synthesis and Characterization of [H2B(3,5-(CF3)2Pz)2]Cu(PPh3)2 Displaying an Unusual Coordination Mode for a Poly(pyrazolyl)borate Ligand. Inorg. Chem. 2000, 39, 2246–2248. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Kuwata, S.; Ikariya, T. Synthesis and Reactivity of Tris(7-azaindolyl)boratoruthenium Complex. Comparison with Poly(methimazolyl)borate Analogue. Chem. Let. 2006, 35, 1224–1225. [Google Scholar] [CrossRef]
- Lenczyk, C.; Roy, D.K.; Ghosh, B.; Schwarzmann, J.; Phukan, A.K.; Braunschweig, H. First Bis(σ)-borane Complexes of Group 6 Transition Metals: Experimental and Theoretical Studies. Chem. Eur. J. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Trofimenko, S. Boron-Pyrazole Chemistry. J. Am. Chem. Soc. 1966, 88, 1842–1844. [Google Scholar] [CrossRef]
- Trofimenko, S. Scorpionates: The Coordination of Poly(pyrazolyl)borate Ligands; Imperial College Press: London, UK, 1999. [Google Scholar] [CrossRef]
- Trofimenko, S. Scorpionates: Genesis, milestones, prognosis. Polyhedron 2004, 23, 197–203. [Google Scholar] [CrossRef]
- Pettinari, C. Scorpionates II: Chelating Borate Ligands; Imperial College Press: London, UK, 2008. [Google Scholar] [CrossRef]
- Yap, G.P.A. A brief history of scorpionates. Acta Cryst. 2013, C69, 937–938. [Google Scholar] [CrossRef] [PubMed]
- Many special issues dedicated to the chemistry of scorpionate ligands has been published; see for example, Pettinari, C. Scorpionate Compounds. Eur. J. Inorg. Chem. 2016, 2016, 2209–2211. [CrossRef]
- Spicer, M.D.; Reglinski, J. Soft Scorpionate Ligands Based on Imidazole-2-thione Donors. Eur. J. Inorg. Chem. 2009, 1553–1574. [Google Scholar] [CrossRef]
- Hill, A.F.; Owen, G.R.; White, A.J.P.; Williams, D.J. The Sting of the Scorpion: A Metallaboratrane. Angew. Chem. Int. Ed. 1999, 38, 2759–2761. [Google Scholar] [CrossRef]
- Owen, G.R. Hydrogen atom storage upon Z-class borane ligand functions: An alternative approach to ligand cooperation. Chem. Soc. Rev. 2012, 41, 3535–3546. [Google Scholar] [CrossRef] [PubMed]
- Owen, G.R. Functional group migrations between boron and metal centres within transition metal–borane and –boryl complexes and cleavage of H–H, E–H and E–E′ bonds. Chem. Commun. 2016, 52, 10712–10726. [Google Scholar] [CrossRef] [PubMed]
- Bouhadir, G.; Bourissou, D. Complexes of ambiphilic ligands: Reactivity and catalytic applications. Chem. Soc. Rev. 2016, 45, 1065–1079. [Google Scholar] [CrossRef]
- Crossley, I.R.; Hill, A.F.; Willis, A.C. Unlocking the metallaboratrane cage: Reversible B–H activation in platinaboratranes. Dalton Trans. 2008, 201–203. [Google Scholar] [CrossRef]
- Neshat, A.; Shahsavari, H.R.; Mastrorilli, P.; Todisco, S.; Haghighi, M.G.; Notash, B. A Borane. A Borane Platinum Complex Undergoing Reversible Hydride Migration in Solution. Inorg. Chem. 2018, 57, 1398–1407. [Google Scholar] [CrossRef]
- Garner, M.; Reglinski, J.; Cassidy, I.; Spicer, M.D.; Kennedy, A.R. Hydrotris(methimazolyl)borate, a soft analogue of hydrotris(pyrazolyl)borate. Preparation and crystal structure of a novel zinc complex. Chem. Commun. 1996, 1975–1976. [Google Scholar] [CrossRef]
- Ma, C.; Hill, A.F. Methimazolyl based diptych bicyclo-[3.3.0]-ruthenaboratranes. Dalton Trans. 2019, 48, 1976–1992. [Google Scholar] [CrossRef] [PubMed]
- Foreman, M.R.S.-J.; Hill, A.F.; Ma, C.; Tshabang, N.; Whited, A.J.P. Synthesis and ligand substitution reactions of κ4-B,S,S′,S′′-ruthenaboratranes. Dalton Trans. 2019, 48, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Schwich, T.; Xiong, Y. 5-Mercaptotetrazolyl-derived metallaboratranes. Dalton Trans. 2019, 48, 2367–2376. [Google Scholar] [CrossRef] [PubMed]
- Gomosta, S.; Ramalakshmi, R.; Arivazhagan, C.; Haridas, A.; Raghavendra, B.; Maheswari, K.; Roisnel, T.; Ghosh, S.Z. Synthesis, Structural Characterization, and Theoretical Studies of Silver(I) Complexes of Dihydrobis(2-mercapto-benzothiazolyl) Borate. Anorg. Allg. Chem. 2019, 645, 588–594. [Google Scholar] [CrossRef]
- Song, D.; Jia, W.L.; Wu, G.; Wang, S. Cu(I) and Zn(II) complexes of 7-azaindole-containing scorpionates: Structures, luminescence and fluxionality. Dalton Trans. 2005, 433–438. [Google Scholar] [CrossRef]
- Wagler, J.; Hill, A.F. 7-Azaindol-7-ylborate: A Novel Bidentate N^BH3 Chelating Ligand. Organometallics 2008, 27, 2350–2353. [Google Scholar] [CrossRef]
- Da Costa, R.C.; Rawe, B.W.; Tsoureas, N.; Haddow, M.F.; Sparkes, H.A.; Tizzard, G.J.; Coles, S.J.; Owen, G.R. Preparation and reactivity of rhodium and iridium complexes containing a methylborohydride based unit supported by two 7-azaindolyl heterocycles. Dalton Trans. 2018, 47, 11047–11057. [Google Scholar] [CrossRef] [Green Version]
- Tsoureas, N.; Hamilton, A.; Haddow, M.F.; Harvey, J.N.; Orpen, A.G.; Owen, G.R. Insight into the Hydrogen Migration Processes Involved in the Formation of Metal–Borane Complexes: Importance of the Third Arm of the Scorpionate Ligand. Organometallics 2013, 32, 2840–2856. [Google Scholar] [CrossRef]
- Holler, S.; Tüchler, M.; Belaj, F.; Veiros, L.F.; Kirchner, K.; Mösch-Zanetti, N.C. Thiopyridazine-Based Copper Boratrane Complexes Demonstrating the Z-type Nature of the Ligand. Inorg. Chem. 2016, 55, 4980–4991. [Google Scholar] [CrossRef]
- Tüchler, M.; Ramböck, M.; Glanzer, S.; Zangger, K.; Belaj, F.; Mösch-Zanetti, N.C. Mono- and Hexanuclear Zinc Halide Complexes with Soft Thiopyridazine Based Scorpionate Ligands. Inorganics 2019, 7, 24. [Google Scholar] [CrossRef]
- Maria, L.; Paulo, A.; Santos, I.C.; Santos, I.; Kurz, P.; Springler, B.; Alberto, R. Very Small and Soft Scorpionates: Water Stable Technetium Tricarbonyl Complexes Combining a Bis-agostic (k3-H, H, S) Binding Motif with Pendant and Integrated Bioactive Molecules. J. Am. Chem. Soc. 2006, 128, 14590–14598. [Google Scholar] [CrossRef] [PubMed]
- Videira, M.; Maria, L.; Paulo, A.; Santos, I.C.; Santos, I.; Vaz, P.D.; Calhorda, M.J. Mixed-Ligand Rhenium Tricarbonyl Complexes Anchored on a (κ2-H,S) Trihydro(mercaptoimidazolyl)borate: A Missing Binding Motif for Soft Scorpionates. Organometallics 2008, 27, 1334–1337. [Google Scholar] [CrossRef]
- Dyson, G.; Hamilton, A.; Mitchell, B.; Owen, G.R. A new family of flexible scorpionate ligands based on 2-mercaptopyridine. Dalton Trans. 2009, 6120–6126. [Google Scholar] [CrossRef] [PubMed]
- Iannetelli, A.; Tizzard, G.J.; Coles, S.J.; Owen, G.R. Sequential Migrations between Boron and Rhodium Centers: A Cooperative Process between Rhodium and a Monosubstituted Borohydride Unit. Inorg. Chem. 2018, 57, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Iannetelli, A.; Tizzard, G.J.; Coles, S.J.; Owen, G.R. Synthesis and Characterization of Platinum and Palladium Complexes Featuring a Rare Secondary Borane Pincer Motif. Organometallics 2018, 37, 2177–2187. [Google Scholar] [CrossRef] [Green Version]
- Zech, A.; Haddow, M.F.; Othman, H.; Owen, G.R. Utilizing the 8-Methoxycyclooct-4-en-1-ide Unit As a Hydrogen Atom Acceptor en Route to “Metal–Borane Pincers”. Organometallics 2012, 31, 6753–6760. [Google Scholar] [CrossRef]
- Anju, R.S.; Roy, D.K.; Mondal, B.; Yuvaraj, K.; Arivazhagan, C.; Saha, K.; Varghese, B.; Ghosh, S. Reactivity of Diruthenium and Dirhodium Analogues of Pentaborane(9): Agostic versus Boratrane Complexes. Angew. Chem. Int. Ed. 2014, 53, 2873–2877. [Google Scholar] [CrossRef]
- Roy, D.K.; Mondal, B.; Anju, R.S.; Ghosh, S. Chemistry of Diruthenium and Dirhodium Analogues of Pentaborane(9): Synthesis and Characterization of Metal N,S-Heterocyclic Carbene and B-Agostic Complexes. Chem. Eur. J. 2015, 21, 3640–3648. [Google Scholar] [CrossRef]
- Anju, R.S.; Mondal, B.; Saha, K.; Panja, S.; Varghese, B.; Ghosh, S. Hydroboration of Alkynes with Zwitterionic Ruthenium–Borate Complexes: Novel Vinylborane Complexes. Chem. Eur. J. 2015, 21, 11393–11400. [Google Scholar] [CrossRef]
- Ramalakshmi, R.; Saha, K.; Roy, D.K.; Varghese, B.; Phukan, A.K.; Ghosh, S. New Routes to a Series of σ-Borane/Borate Complexes of Molybdenum and Ruthenium. Chem. Eur. J. 2015, 21, 17191–17195. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.K.; Borthakur, R.; De, A.; Varghese, B.; Phukan, A.K.; Ghosh, S. Synthesis and Characterization of Bis(sigma)borate and Bis–zwitterionic Complexes of Rhodium and Iridium. Chem. Select 2016, 1, 3757–3761. [Google Scholar] [CrossRef]
- Saha, K.; Joseph, B.; Ramalakshmi, R.; Anju, R.S.; Varghese, B.; Ghosh, S. η4-HBCC-σ,π-Borataallyl Complexes of Ruthenium Comprising an Agostic Interaction. Chem. Eur. J. 2016, 22, 7871–7878. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Joseph, B.; Borthakur, R.; Ramalakshmi, R.; Roisnel, T.; Ghosh, S. Chemistry of ruthenium σ-borane complex, [Cp∗RuCO(μ-H)BH2L] (Cp∗ = η5-C5Me5; L = C7H4NS2) with terminal and internal alkynes: Structural characterization of vinyl hydroborate and vinyl complexes of ruthenium. Polyhedron 2017, 125, 246–252. [Google Scholar] [CrossRef]
- Nako, A.E.; White, A.J.P.; Crimmin, M.R. Bis(σ-B–H) complexes of copper(I): Precursors to a heterogeneous amine–borane dehydrogenation catalyst. Dalton Trans. 2015, 44, 12530–12534. [Google Scholar] [CrossRef] [PubMed]
- Hicken, A.; White, A.J.P.; Crimmin, M.R. Reversible Coordination of Boron–, Aluminum–, Zinc–, Magnesium–, and Calcium–Hydrogen Bonds to Bent {CuL2} Fragments: Heavy σ Complexes of the Lightest Coinage Metal. Inorg. Chem. 2017, 56, 8669–8682. [Google Scholar] [CrossRef] [PubMed]
- This signal was unambiguously confirmed via a COSY NMR experiment
- Coles, S.J.; Gale, P.A. Changing and challenging times for service crystallography. Chem. Sci. 2012, 3, 683. [Google Scholar] [CrossRef]
- CrysAlisPro Software System, Rigaku, V1.171.40.40a, Rigaku Oxford Diffraction, 2019
- Sheldrick, G.M. Crystal structure refinement with ShelXL. Acta Cryst. 2015, C27, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. Acta Cryst. 2015, A71, 3. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
Figure 1.
Selected examples of ligands in which the borohydride unit is positioned away from the metal center (left), directed towards the metal center (middle), and a system in which a B‒H activation has occurred (right). The additional atom between boron and the donor atoms is typically necessary for metal‒boron bond formation.
Figure 1.
Selected examples of ligands in which the borohydride unit is positioned away from the metal center (left), directed towards the metal center (middle), and a system in which a B‒H activation has occurred (right). The additional atom between boron and the donor atoms is typically necessary for metal‒boron bond formation.

Figure 2.
The monosubstituted borohydride salt, Na[H3B(mp)].

Scheme 1.
Synthesis of [Cu{κ3-S,H,H-H3B(mp)}(PR3)] (R = Ph, 1; Cy, 2).

Figure 3.
Molecular structures of [Cu{κ3-S,H,H-H3B(mp)}(PR3)] (R = Ph, 1; Cy, 2). Thermal ellipsoids drawn at 50% level. Hydrogen atoms, with the exception of those attached to the boron centers, have been omitted for clarity. Both structures contain disorder in the position of the [H3B(mp)]− ligand. Only the major component is shown (see text for details).
Figure 3.
Molecular structures of [Cu{κ3-S,H,H-H3B(mp)}(PR3)] (R = Ph, 1; Cy, 2). Thermal ellipsoids drawn at 50% level. Hydrogen atoms, with the exception of those attached to the boron centers, have been omitted for clarity. Both structures contain disorder in the position of the [H3B(mp)]− ligand. Only the major component is shown (see text for details).

Figure 4.
Two bonding descriptions for the [H3B(mp)]−.

Figure 5.
Numbering Scheme used for [H3B(mp)]− and PCy3.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected NMR (ppm) and IR (cm−1) spectroscopic data for [HnB(mp)4−n] pro-ligands and their corresponding copper complexes.
Table 1.
Selected NMR (ppm) and IR (cm−1) spectroscopic data for [HnB(mp)4−n] pro-ligands and their corresponding copper complexes.
| Compound 1 | 11B{1H} NMR 2 | 31P{1H} NMR | 13C{1H} NMR C=S | 1H{11B} NMR 3 BHn | IR B–H 4 |
|---|---|---|---|---|---|
| Na[H3B(mp)] 5 | −14.1 (44) | - | 181.3 | 2.11 | 2307 |
| [Cu{H3B(mp)}(PPh3)] (1) | −13.9 (113) | 4.8 | 175.9 | 2.64 | 2439 (t)/2078 (κ2) |
| [Cu{H3B(mp)}(PCy3)] (2) | −13.4 (90) | 27.2 | 176.1 | 2.42 | 2448 (t)/2085 (κ2) |
| Na[H2B(mp)2] 6 | −3.7 (211) | - | 182.6 | 3.64 | 2438, 2370 |
| [Cu{H2B(mp)2}(PPh3)] (3) | 0.7 (265) | 1.7 | n.o. 7 | 4.12 | 2425 |
| [Cu{H2B(mp)2}(PCy3)] | −0.7 (248) | 19.0 | 178.2 | 3.99 | 2374 |
| K[HB(mp)3] 6 | 4.4 (560) | - | 182.5 | 4.83 | 2468 |
| [Cu{HB(mp)3}(PPh3)] | −0.1 (412) | −2.4 | 178.3 | n.o. 7 | 2458 |
| [Cu{HB(mp)3}(PCy3)] | −0.5 (331) | 17.4 | 181.0 | 5.86 | n.o. 7 |
Note: 1 The NMR spectroscopic data for all complexes were recorded in CDCl3; 2 the values in brackets are the half-height widths of the measurement of the signal; 3 this signal corresponds to chemical environments of hydrogen substituents at the boron center. In all cases, only one single chemical environment was observed for the BHn units; 4 recorded as a powder film, where clear the terminal (t) and BH2Cu bridging modes (κ2) are highlighted in brackets; 5 in CD3CN NMR solvent; 6 in DMSO-d6 NMR solvent; 7 this chemical environment or B–H stretch was not observed in this spectrum.
Table 2.
Selected Bond Distances (Å) and Angles (°) for 1–3.
| [Cu{H3B(mp)}PPh3] 1 | [Cu{H3B(mp)}PCy3] 2 | [Cu{H2B(mp)2}PPh3] 3 | |
|---|---|---|---|
| Cu(1)–P(1) | 2.1789(4) | 2.1876(4) | 2.216(3) |
| Cu(1)–B(1) | 2.113(17)/2.229(14) | 2.153(16)/2.10(3) | 2.7479(15) |
| Cu(1)–S(1) | 2.205(2)/2.221(4) | 2.2523(12)/2.296(12) | 2.255(4) and 2.248(4) |
| C(1)–S(1) | 1.7515(17)/1.722(2) | 1.7244(17)/1.751(13) | 1.707(14) and 1.708(14) |
| B(1)–N(1) | 1.551(8)/1.465(10) | 1.602(16)/1.61(2) | 1.592(2) and 1.583(18) |
| N(1)–C(1) | 1.3506(19)/1.3506(19) | 1.3550(19)/1.3550(19) | 1.3649(17) and 1.3648(19) |
| B(1)–H(1AA) | 1.17(2)/1.18(2) | 1.16(2)/1.16(2) | - |
| B(1)–H(1AB) | 1.16(2)/1.18(2) | 1.17(2)/1.15(2) | 1.090(18) (terminal) |
| B(1)–H(1AC) | 1.17(2)/1.17(2) | 1.14(2)/1.15(2) | 1.150(17) (bridging) |
| Cu(1)–H(1AA) | 1.75(3)/1.81(4) | 1.75(2)/1.68(8) | 1.832(17) |
| Cu(1)–H(1AB) | 1.81(3)/1.85(4) | 1.81(2)/1.82(8) | - |
| S(1)–Cu(1)–P(1) | 129.93(3)/134.69(5) | 129.93(3)/135.9(3) | 111.88(15) and 124.56(14) |
| S(1)–Cu(1)–B(1) | 89.2(2)/87.3(2) | 89.7(4)/90.2(5) | 82.29(3) and 80.27(3) |
| P(1)–Cu(1)–B(1) | 140.5(2)/137.5(3) | 140.3(4)/133.9(6) | 135.64(3) |
| Σangles around Cu 4 | 359.63/359.49 | 359.93/360.0 | 350.4 |
| C(1)–S(1)–Cu(1) | 99.53(9)/99.14(16) | 99.53(8)/96.2(5) | 106.49(5) and 109.83(5) |
| N(1)–B(1)–Cu(1) | 110.0(8)/108.7(7) | 107.0(8)/110.3(13) | 95.36 and 99.09 |
Note: 1 the [H3B(mp)]− ligand is disordered over two positions (with an approximate ratio 56:44). Where a second value is provided in the table, it represents the value corresponding to the minor occupancy component; 2 the [H3B(mp)]− ligand is disordered over two positions (with an approximate ratio 79:21). Where a second values is provided in the table, it represents the value corresponding to the minor occupancy component; 3 data obtained from reference [36], the two values here result from the fact that there are two mp units within the complex; 4 the value quoted involves the sum of all angles around the copper center involving all non-hydrogen atoms.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Goldsworthy, J.; Thomas, S.D.; Tizzard, G.J.; Coles, S.J.; Owen, G.R. Adding to the Family of Copper Complexes Featuring Borohydride Ligands Based on 2-Mercaptopyridyl Units. Inorganics 2019, 7, 93. https://doi.org/10.3390/inorganics7080093
AMA Style
Goldsworthy J, Thomas SD, Tizzard GJ, Coles SJ, Owen GR. Adding to the Family of Copper Complexes Featuring Borohydride Ligands Based on 2-Mercaptopyridyl Units. Inorganics. 2019; 7(8):93. https://doi.org/10.3390/inorganics7080093
Chicago/Turabian StyleGoldsworthy, Joseph, Simon D. Thomas, Graham J. Tizzard, Simon J. Coles, and Gareth R. Owen. 2019. "Adding to the Family of Copper Complexes Featuring Borohydride Ligands Based on 2-Mercaptopyridyl Units" Inorganics 7, no. 8: 93. https://doi.org/10.3390/inorganics7080093
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.